Cell line analysis
George Howitt
2023-05-22
Last updated: 2023-07-05
Checks: 7 0
Knit directory: hashtag-demux-paper/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20230522) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 2e25259. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/BAL_data/.DS_Store
Untracked files:
Untracked: .ipynb_checkpoints/
Untracked: Dear editors.docx
Untracked: NAR.cls
Untracked: analysis/Fscore_MCC_comparison.Rmd
Untracked: analysis/gmm_demux.Rmd
Untracked: analysis/hashsolo_prep.Rmd
Untracked: analysis/run_GMM_demux_BAL.sh
Untracked: cover_letter.docx
Untracked: data/BAL_data/batch1_all_methods.SEU.rds
Untracked: data/BAL_data/batch1_c1_donors_original.csv
Untracked: data/BAL_data/batch1_c1_hto_counts_original.csv
Untracked: data/BAL_data/batch1_c1_relabelled.SEU.rds
Untracked: data/BAL_data/batch1_c2_donors_original.csv
Untracked: data/BAL_data/batch1_c2_hto_counts_original.csv
Untracked: data/BAL_data/batch1_c2_relabelled.SEU.rds
Untracked: data/BAL_data/batch1_relabelled.SEU.rds
Untracked: data/BAL_data/batch2_all_methods.SEU.rds
Untracked: data/BAL_data/batch2_c1_donors_original.csv
Untracked: data/BAL_data/batch2_c1_hto_counts_original.csv
Untracked: data/BAL_data/batch2_c1_relabelled.SEU.rds
Untracked: data/BAL_data/batch2_c2_donors_original.csv
Untracked: data/BAL_data/batch2_c2_hto_counts_original.csv
Untracked: data/BAL_data/batch2_c2_relabelled.SEU.rds
Untracked: data/BAL_data/batch2_relabelled.SEU.rds
Untracked: data/BAL_data/batch3_all_methods.SEU.rds
Untracked: data/BAL_data/batch3_c1_donors_original.csv
Untracked: data/BAL_data/batch3_c1_hto_counts_original.csv
Untracked: data/BAL_data/batch3_c1_relabelled.SEU.rds
Untracked: data/BAL_data/batch3_c2_donors_original.csv
Untracked: data/BAL_data/batch3_c2_hto_counts_original.csv
Untracked: data/BAL_data/batch3_c2_relabelled.SEU.rds
Untracked: data/BAL_data/batch3_relabelled.SEU.rds
Untracked: data/adata/batch1_c1_hashsolo.csv
Untracked: data/adata/batch1_c1_hs_n10_d10.csv
Untracked: data/adata/batch1_c1_hs_n10_d20.csv
Untracked: data/adata/batch1_c1_hs_n10_d30.csv
Untracked: data/adata/batch1_c1_hs_n1_d10.csv
Untracked: data/adata/batch1_c1_hs_n1_d20.csv
Untracked: data/adata/batch1_c1_hs_n1_d30.csv
Untracked: data/adata/batch1_c1_hs_n5_d10.csv
Untracked: data/adata/batch1_c1_hs_n5_d30.csv
Untracked: data/adata/batch1_c2_hashsolo.csv
Untracked: data/adata/batch1_c2_hs_n10_d10.csv
Untracked: data/adata/batch1_c2_hs_n10_d20.csv
Untracked: data/adata/batch1_c2_hs_n10_d30.csv
Untracked: data/adata/batch1_c2_hs_n1_d10.csv
Untracked: data/adata/batch1_c2_hs_n1_d20.csv
Untracked: data/adata/batch1_c2_hs_n1_d30.csv
Untracked: data/adata/batch1_c2_hs_n5_d10.csv
Untracked: data/adata/batch1_c2_hs_n5_d30.csv
Untracked: data/adata/batch2_c1_hashsolo.csv
Untracked: data/adata/batch2_c1_hs_n10_d10.csv
Untracked: data/adata/batch2_c1_hs_n10_d20.csv
Untracked: data/adata/batch2_c1_hs_n10_d30.csv
Untracked: data/adata/batch2_c1_hs_n1_d10.csv
Untracked: data/adata/batch2_c1_hs_n1_d20.csv
Untracked: data/adata/batch2_c1_hs_n1_d30.csv
Untracked: data/adata/batch2_c1_hs_n5_d10.csv
Untracked: data/adata/batch2_c1_hs_n5_d30.csv
Untracked: data/adata/batch2_c2_hashsolo.csv
Untracked: data/adata/batch2_c2_hs_n10_d10.csv
Untracked: data/adata/batch2_c2_hs_n10_d20.csv
Untracked: data/adata/batch2_c2_hs_n10_d30.csv
Untracked: data/adata/batch2_c2_hs_n1_d10.csv
Untracked: data/adata/batch2_c2_hs_n1_d20.csv
Untracked: data/adata/batch2_c2_hs_n1_d30.csv
Untracked: data/adata/batch2_c2_hs_n5_d10.csv
Untracked: data/adata/batch2_c2_hs_n5_d30.csv
Untracked: data/adata/batch3_c1_hashsolo.csv
Untracked: data/adata/batch3_c1_hs_n10_d10.csv
Untracked: data/adata/batch3_c1_hs_n10_d20.csv
Untracked: data/adata/batch3_c1_hs_n10_d30.csv
Untracked: data/adata/batch3_c1_hs_n1_d10.csv
Untracked: data/adata/batch3_c1_hs_n1_d20.csv
Untracked: data/adata/batch3_c1_hs_n1_d30.csv
Untracked: data/adata/batch3_c1_hs_n5_d10.csv
Untracked: data/adata/batch3_c1_hs_n5_d30.csv
Untracked: data/adata/batch3_c2_hashsolo.csv
Untracked: data/adata/batch3_c2_hs_n10_d10.csv
Untracked: data/adata/batch3_c2_hs_n10_d20.csv
Untracked: data/adata/batch3_c2_hs_n10_d30.csv
Untracked: data/adata/batch3_c2_hs_n1_d10.csv
Untracked: data/adata/batch3_c2_hs_n1_d20.csv
Untracked: data/adata/batch3_c2_hs_n1_d30.csv
Untracked: data/adata/batch3_c2_hs_n5_d10.csv
Untracked: data/adata/batch3_c2_hs_n5_d30.csv
Untracked: data/adata/solid_tissue_batch1_hashsolo.csv
Untracked: data/adata/solid_tissue_batch2_hashsolo.csv
Untracked: data/solid_tumor_data/
Untracked: figures/QC_plots_new.png
Untracked: figures/Users/
Untracked: filter_wrong_empties.Rmd
Untracked: iscb_long_abstract.docx
Untracked: iscb_long_abstract.pdf
Untracked: oup-authoring-template/
Untracked: output/mean_fscore_mcc.xlsx
Untracked: paper_latex/
Unstaged changes:
Modified: analysis/index.Rmd
Modified: data/.DS_Store
Deleted: data/GMM-Demux/SSD_mtx/barcodes.tsv.gz
Deleted: data/GMM-Demux/SSD_mtx/features.tsv.gz
Deleted: data/GMM-Demux/SSD_mtx/matrix.mtx.gz
Deleted: data/GMM-Demux/batch1_c1_hto_counts_transpose.csv
Deleted: data/GMM-Demux/batch1_c2_hto_counts_transpose.csv
Deleted: data/GMM-Demux/batch2_c1_hto_counts_transpose.csv
Deleted: data/GMM-Demux/batch2_c2_hto_counts_transpose.csv
Deleted: data/GMM-Demux/batch3_c1_hto_counts_transpose.csv
Deleted: data/GMM-Demux/batch3_c2_hto_counts_transpose.csv
Deleted: data/GMM-Demux/gmm_out_LMO_c1/full_report/GMM_full.config
Deleted: data/GMM-Demux/gmm_out_LMO_c1/full_report/GMM_full.csv
Deleted: data/GMM-Demux/gmm_out_LMO_c1/simplified_report/GMM_simplified.config
Deleted: data/GMM-Demux/gmm_out_LMO_c1/simplified_report/GMM_simplified.csv
Deleted: data/GMM-Demux/gmm_out_LMO_c2/full_report/GMM_full.config
Deleted: data/GMM-Demux/gmm_out_LMO_c2/full_report/GMM_full.csv
Deleted: data/GMM-Demux/gmm_out_LMO_c2/simplified_report/GMM_simplified.config
Deleted: data/GMM-Demux/gmm_out_LMO_c2/simplified_report/GMM_simplified.csv
Deleted: data/GMM-Demux/gmm_out_LMO_c3/full_report/GMM_full.config
Deleted: data/GMM-Demux/gmm_out_LMO_c3/full_report/GMM_full.csv
Deleted: data/GMM-Demux/gmm_out_LMO_c3/simplified_report/GMM_simplified.config
Deleted: data/GMM-Demux/gmm_out_LMO_c3/simplified_report/GMM_simplified.csv
Deleted: data/GMM-Demux/gmm_out_batch1_c1/full_report/GMM_full.config
Deleted: data/GMM-Demux/gmm_out_batch1_c1/full_report/GMM_full.csv
Deleted: data/GMM-Demux/gmm_out_batch1_c1/simplified_report/GMM_simplified.config
Deleted: data/GMM-Demux/gmm_out_batch1_c1/simplified_report/GMM_simplified.csv
Deleted: data/GMM-Demux/gmm_out_batch1_c2/full_report/GMM_full.config
Deleted: data/GMM-Demux/gmm_out_batch1_c2/full_report/GMM_full.csv
Deleted: data/GMM-Demux/gmm_out_batch1_c2/simplified_report/GMM_simplified.config
Deleted: data/GMM-Demux/gmm_out_batch1_c2/simplified_report/GMM_simplified.csv
Deleted: data/GMM-Demux/gmm_out_batch2_c1/full_report/GMM_full.config
Deleted: data/GMM-Demux/gmm_out_batch2_c1/full_report/GMM_full.csv
Deleted: data/GMM-Demux/gmm_out_batch2_c1/simplified_report/GMM_simplified.config
Deleted: data/GMM-Demux/gmm_out_batch2_c1/simplified_report/GMM_simplified.csv
Deleted: data/GMM-Demux/gmm_out_batch2_c2/full_report/GMM_full.config
Deleted: data/GMM-Demux/gmm_out_batch2_c2/full_report/GMM_full.csv
Deleted: data/GMM-Demux/gmm_out_batch2_c2/simplified_report/GMM_simplified.config
Deleted: data/GMM-Demux/gmm_out_batch2_c2/simplified_report/GMM_simplified.csv
Deleted: data/GMM-Demux/gmm_out_batch3_c1/full_report/GMM_full.config
Deleted: data/GMM-Demux/gmm_out_batch3_c1/full_report/GMM_full.csv
Deleted: data/GMM-Demux/gmm_out_batch3_c1/simplified_report/GMM_simplified.config
Deleted: data/GMM-Demux/gmm_out_batch3_c1/simplified_report/GMM_simplified.csv
Deleted: data/GMM-Demux/gmm_out_batch3_c2/full_report/GMM_full.config
Deleted: data/GMM-Demux/gmm_out_batch3_c2/full_report/GMM_full.csv
Deleted: data/GMM-Demux/gmm_out_batch3_c2/simplified_report/GMM_simplified.config
Deleted: data/GMM-Demux/gmm_out_batch3_c2/simplified_report/GMM_simplified.csv
Deleted: data/GMM-Demux/lmo_counts_capture1_transpose.csv
Deleted: data/GMM-Demux/lmo_counts_capture2_transpose.csv
Deleted: data/GMM-Demux/lmo_counts_capture3_transpose.csv
Deleted: data/GMM-Demux/run_GMM_demux_BAL.sh
Deleted: data/GMM-Demux/run_GMM_demux_LMO.sh
Modified: data/adata/batch1_HTOs.csv
Modified: data/adata/batch1_c1_barcodes.csv
Modified: data/adata/batch1_c1_counts.mtx
Modified: data/adata/batch1_c2_barcodes.csv
Modified: data/adata/batch1_c2_counts.mtx
Modified: data/adata/batch2_HTOs.csv
Modified: data/adata/batch2_c1_barcodes.csv
Modified: data/adata/batch2_c1_counts.mtx
Modified: data/adata/batch2_c2_barcodes.csv
Modified: data/adata/batch2_c2_counts.mtx
Modified: data/adata/batch3_HTOs.csv
Modified: data/adata/batch3_c1_barcodes.csv
Modified: data/adata/batch3_c1_counts.mtx
Modified: data/adata/batch3_c2_barcodes.csv
Modified: data/adata/batch3_c2_counts.mtx
Deleted: data/batch1_c1_donors.csv
Deleted: data/batch1_c1_hto_counts.csv
Deleted: data/batch1_c2_donors.csv
Deleted: data/batch1_c2_hto_counts.csv
Deleted: data/batch1_hto_counts.csv
Deleted: data/batch2_c1_donors.csv
Deleted: data/batch2_c1_hto_counts.csv
Deleted: data/batch2_c2_donors.csv
Deleted: data/batch2_c2_hto_counts.csv
Deleted: data/batch2_hto_counts.csv
Deleted: data/batch3_c1_donors.csv
Deleted: data/batch3_c1_hto_counts.csv
Deleted: data/batch3_c2_donors.csv
Deleted: data/batch3_c2_hto_counts.csv
Deleted: data/batch3_hto_counts.csv
Deleted: data/lmo_counts.csv
Deleted: data/lmo_counts_capture1.csv
Deleted: data/lmo_counts_capture2.csv
Deleted: data/lmo_counts_capture3.csv
Deleted: data/lmo_donors.csv
Deleted: data/lmo_donors_capture1.csv
Deleted: data/lmo_donors_capture2.csv
Deleted: data/lmo_donors_capture3.csv
Modified: figures/QC_plots.png
Modified: figures/category_fractions.png
Modified: hashsolo_calls.ipynb
Modified: notebook_for_paper.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/cell_line_analysis.Rmd)
and HTML (docs/cell_line_analysis.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 2e25259 | George Howitt | 2023-07-05 | wflow_publish("analysis/cell_line_analysis.Rmd") |
#Load libraries
suppressPackageStartupMessages({
library(here)
library(BiocStyle)
library(dplyr)
library(janitor)
library(ggplot2)
library(cowplot)
library(patchwork)
library(DropletUtils)
library(tidyverse)
library(scuttle)
library(scater)
library(Seurat)
library(pheatmap)
library(speckle)
library(dittoSeq)
library(cellhashR)
library(RColorBrewer)
library(demuxmix)
library(ComplexHeatmap)
library(tidyHeatmap)
library(viridis)
}
)Cell line data.
This notebook contains the analysis code for all results and figures relating to the cell line data set in the paper “Benchmarking single-cell hashtag oligo demultiplexing methods”.
The data set consists of one batch with three genetically distinct samples in each. The batch was processed in three separate captures. We run all the demultiplexing methods on a per-capture level before then recombining for later analysis.
Data loading and reduction.
Load in counts matrices and genetic IDs.
lmo_counts_c1 <- read.csv(here("data", "cell_line_data", "lmo_counts_capture1.csv"), check.names = FALSE, row.names = 1)
lmo_counts_c2 <- read.csv(here("data", "cell_line_data", "lmo_counts_capture2.csv"), check.names = FALSE, row.names = 1)
lmo_counts_c3 <- read.csv(here("data", "cell_line_data", "lmo_counts_capture3.csv"), check.names = FALSE, row.names = 1)
lmo_donors_c1 <- read.csv(here("data", "cell_line_data", "lmo_donors_capture1.csv"), check.names = FALSE, row.names = 1)
lmo_donors_c2 <- read.csv(here("data", "cell_line_data", "lmo_donors_capture2.csv"), check.names = FALSE, row.names = 1)
lmo_donors_c3 <- read.csv(here("data", "cell_line_data", "lmo_donors_capture3.csv"), check.names = FALSE, row.names = 1)Lists associating the HTOs with the genetic donors.
LMO_list <- c("CL 01", "CL 02", "CL 03", "Doublet", "Negative")
donor_LMO_list <- list("CL A" = "CL 01",
"CL B" = "CL 02",
"CL C" = "CL 03",
"Doublet" = "Doublet",
"Negative" = "Negative")
LMO_donor_list <- list("CL 01" = "CL A",
"CL 02" = "CL B",
"CL 03" = "CL C",
"Doublet" = "Doublet",
"Negative" = "Negative")Create Seurat objects
seu_lmo_c1 <- CreateSeuratObject(counts = lmo_counts_c1, assay = "HTO")
seu_lmo_c2 <- CreateSeuratObject(counts = lmo_counts_c2, assay = "HTO")
seu_lmo_c3 <- CreateSeuratObject(counts = lmo_counts_c3, assay = "HTO")seu_lmo_c1$Barcode <- colnames(seu_lmo_c1)
seu_lmo_c2$Barcode <- colnames(seu_lmo_c2)
seu_lmo_c3$Barcode <- colnames(seu_lmo_c3)Add genetic donor information to Seurat objects
seu_lmo_c1$genetic_donor <- lmo_donors_c1$genetic_donor
seu_lmo_c2$genetic_donor <- lmo_donors_c2$genetic_donor
seu_lmo_c3$genetic_donor <- lmo_donors_c3$genetic_donorMerge together for QC comparison
seu_lmo <- merge(seu_lmo_c1, c(seu_lmo_c2, seu_lmo_c3))Run PCAs and tSNEs
DefaultAssay(seu_lmo_c1) <- "HTO"
seu_lmo_c1 <- NormalizeData(seu_lmo_c1, assay = "HTO", normalization.method = "CLR")Normalizing across featuresseu_lmo_c1 <- ScaleData(seu_lmo_c1, features = rownames(seu_lmo_c1),
verbose = FALSE)
seu_lmo_c1 <- RunPCA(seu_lmo_c1, features = rownames(seu_lmo_c1), approx = FALSE, verbose = FALSE)
#seu_lmo_c1 <- RunTSNE(seu_lmo_c1, dims = 1:3, perplexity = 100, check_duplicates = FALSE, verbose = FALSE)
DefaultAssay(seu_lmo_c2) <- "HTO"
seu_lmo_c2 <- NormalizeData(seu_lmo_c2, assay = "HTO", normalization.method = "CLR")Normalizing across featuresseu_lmo_c2 <- ScaleData(seu_lmo_c2, features = rownames(seu_lmo_c2),
verbose = FALSE)
seu_lmo_c2 <- RunPCA(seu_lmo_c2, features = rownames(seu_lmo_c2), approx = FALSE, verbose = FALSE)
#seu_lmo_c2 <- RunTSNE(seu_lmo_c2, dims = 1:3, perplexity = 100, check_duplicates = FALSE, verbose = FALSE)
DefaultAssay(seu_lmo_c3) <- "HTO"
seu_lmo_c3 <- NormalizeData(seu_lmo_c3, assay = "HTO", normalization.method = "CLR")Normalizing across featuresseu_lmo_c3 <- ScaleData(seu_lmo_c3, features = rownames(seu_lmo_c3),
verbose = FALSE)
seu_lmo_c3 <- RunPCA(seu_lmo_c3, features = rownames(seu_lmo_c3), approx = FALSE, verbose = FALSE)
#seu_lmo_c3 <- RunTSNE(seu_lmo_c3, dims = 1:3, perplexity = 100, check_duplicates = FALSE, verbose = FALSE)DefaultAssay(seu_lmo) <- "HTO"
seu_lmo <- NormalizeData(seu_lmo, assay = "HTO", normalization.method = "CLR")Normalizing across featuresseu_lmo <- ScaleData(seu_lmo, features = rownames(seu_lmo),
verbose = FALSE)
seu_lmo <- RunPCA(seu_lmo, features = rownames(seu_lmo), approx = FALSE, verbose = FALSE)
seu_lmo <- RunTSNE(seu_lmo, dims = 1:3, perplexity = 100, check_duplicates = FALSE, verbose = FALSE)QC plots
Density plots per barcode. In ideal conditions the density of the hashtag counts should appear bimodal, with a lower peak corresponding to the background and the higher peak corresponding to the signal.
df <- as.data.frame(t(seu_lmo[["HTO"]]@counts))
colnames(df) <- LMO_donor_list[colnames(df)]
df %>%
pivot_longer(cols = starts_with("CL")) %>%
mutate(logged = log(value + 1)) %>%
ggplot(aes(x = logged)) +
xlab("log(counts)") +
xlim(0.1,8) +
geom_density(adjust = 2) +
facet_wrap(~name, scales = "fixed", ncol = 3) -> p1
p1Warning: Removed 2647 rows containing non-finite values (`stat_density()`).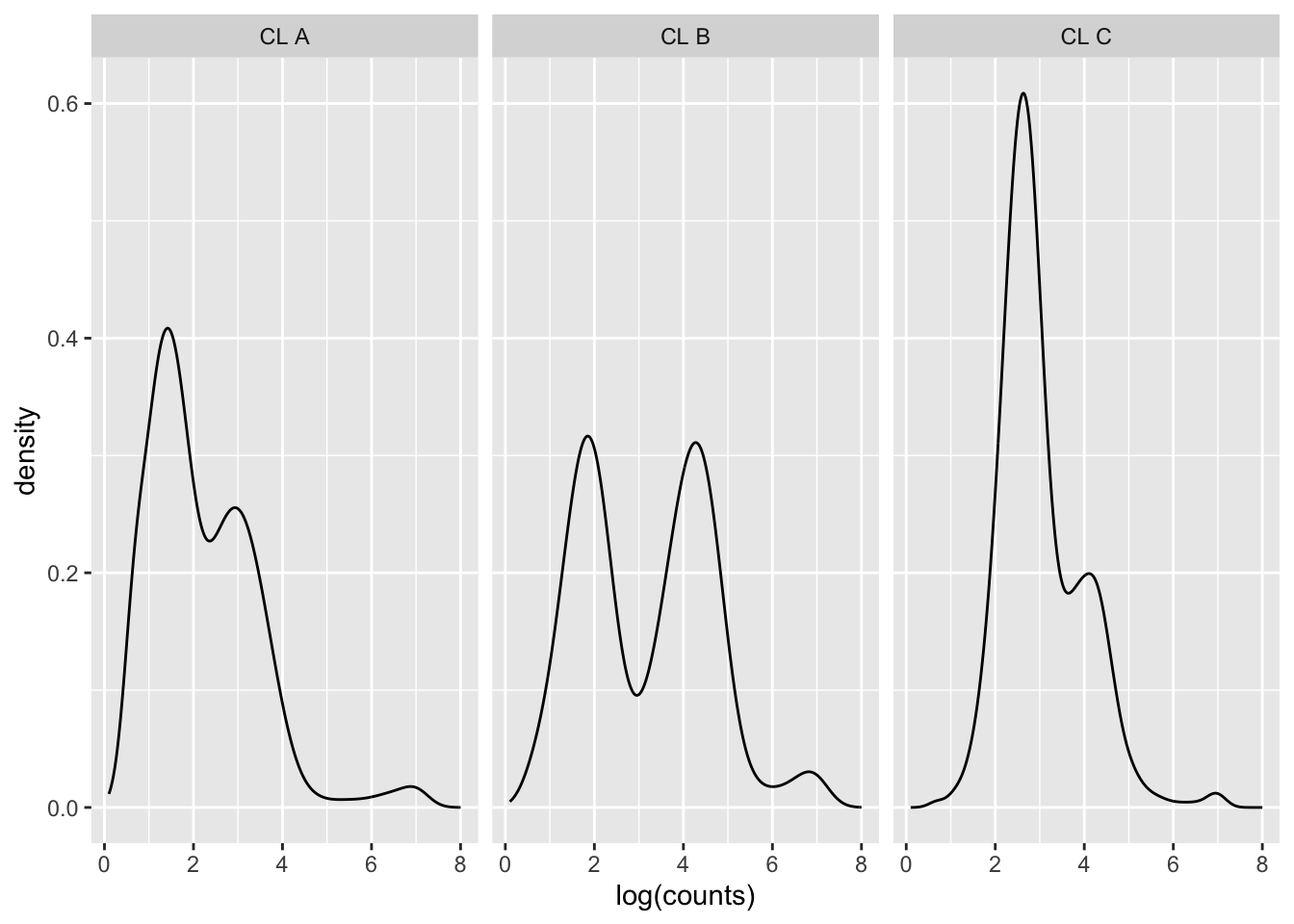
p2 <- DimPlot(seu_lmo, group.by = "genetic_donor") +
theme(axis.text.x = element_blank(), axis.ticks.x = element_blank(),
axis.text.y = element_blank(), axis.ticks.y = element_blank(),
axis.line.x = element_blank(), axis.line.y = element_blank(),
plot.title = element_blank())
p2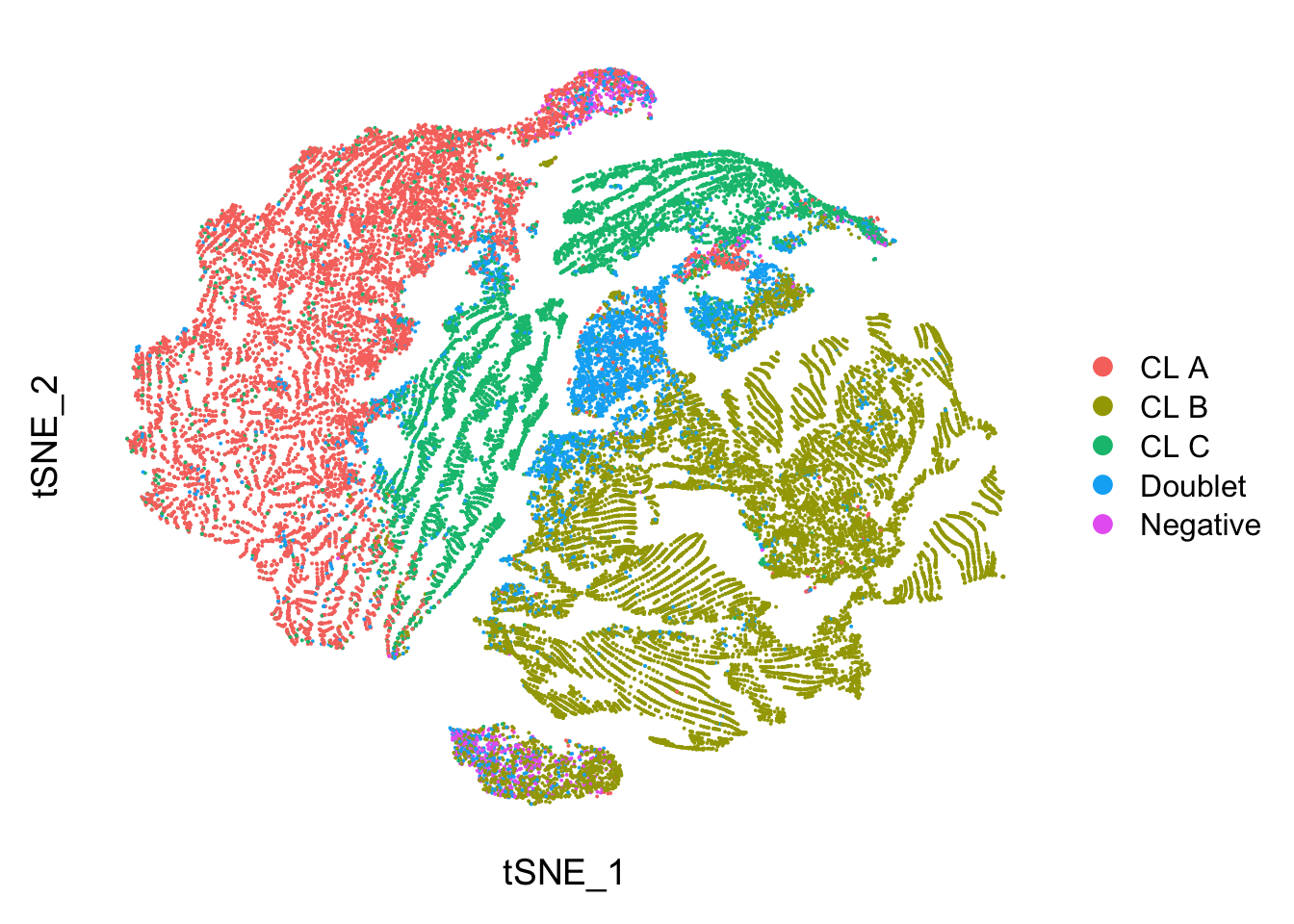
(p1 | p2) + plot_annotation(tag_levels = 'a') &
theme(plot.title = element_text(face = "plain", size = 10),
plot.tag = element_text(face = 'plain'))Warning: Removed 2647 rows containing non-finite values (`stat_density()`).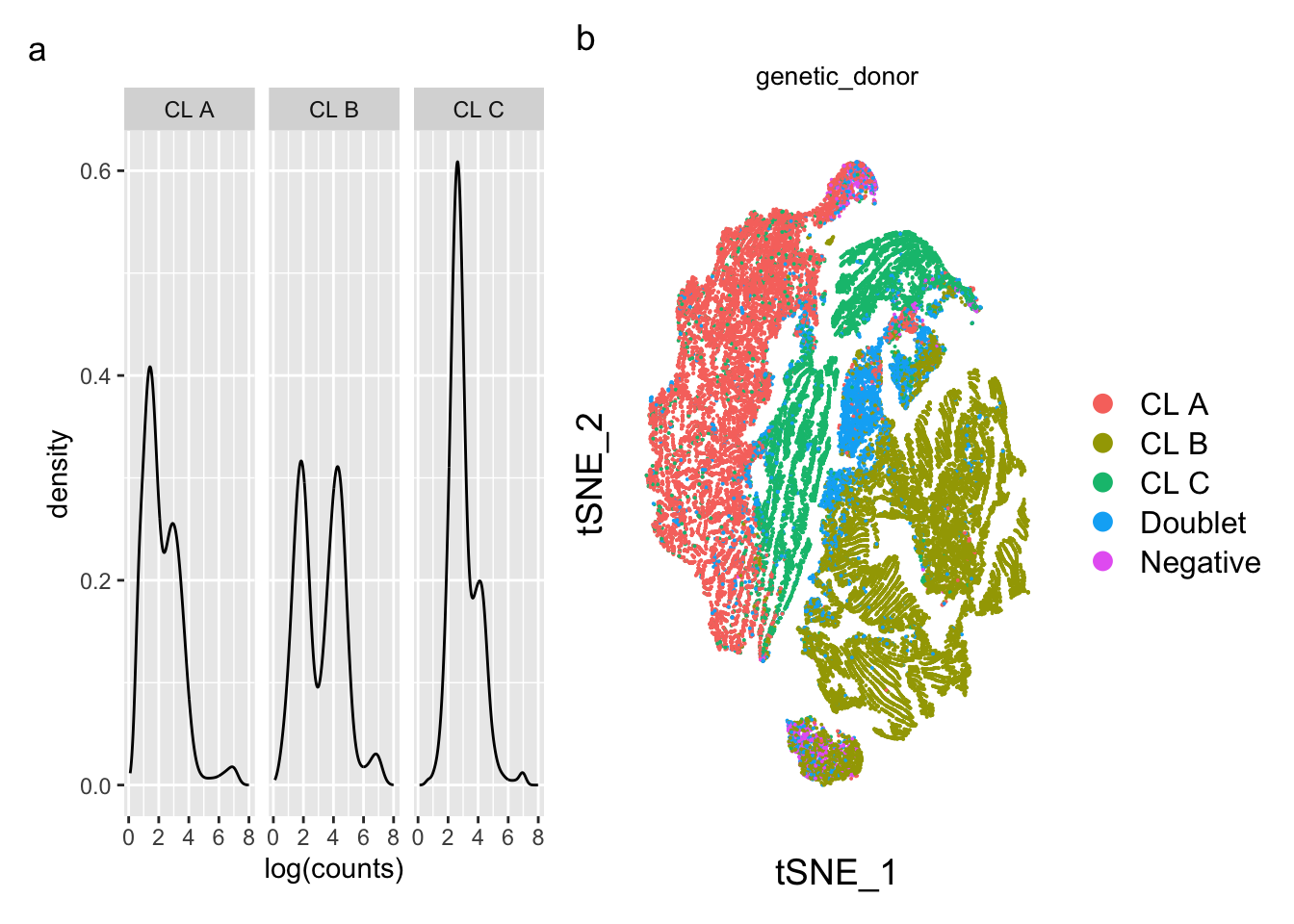
#ggsave("LMO_QC.png",
# plot = (p1 | p2) + plot_annotation(tag_levels = 'a') &
# theme(plot.title = element_text(face = "plain", size = 10),
# plot.tag = element_text(face = 'plain')),
# device = "png",
# path = here("paper_latex", "figures"),
# width = 10, height = 4,
# units = "in",
# dpi = 300)Hashtag-based demultiplexing
hashedDrops
This function creates a list of hashedDrops calls. Its defaults are the same as hashedDrops
create_hashedDrops_factor <- function(seurat_object, confident.min = 2,
doublet.nmads = 3, doublet.min = 2) {
hto_counts <- GetAssayData(seurat_object[["HTO"]], slot = "counts")
hash_stats <- DropletUtils::hashedDrops(hto_counts, confident.min = confident.min,
doublet.nmads = doublet.nmads, doublet.min = doublet.min)
hash_stats$Best <- rownames(seurat_object[["HTO"]])[hash_stats$Best]
hash_stats$Second <- rownames(seurat_object[["HTO"]])[hash_stats$Second]
HTO_assignments <- factor(case_when(
hash_stats$Confident == TRUE ~ hash_stats$Best,
hash_stats$Doublet == TRUE ~ "Doublet",
TRUE ~ "Negative"))
return(HTO_assignments)
}Making factors with “best” parameters
seu_lmo_c1$hashedDrops_calls <- create_hashedDrops_factor(seu_lmo_c1, confident.min = 0.5)
seu_lmo_c2$hashedDrops_calls <- create_hashedDrops_factor(seu_lmo_c2, confident.min = 0.5)
seu_lmo_c3$hashedDrops_calls <- create_hashedDrops_factor(seu_lmo_c3, confident.min = 0.5)Now with default parameters
seu_lmo_c1$hashedDrops_default_calls <- create_hashedDrops_factor(seu_lmo_c1)
seu_lmo_c2$hashedDrops_default_calls <- create_hashedDrops_factor(seu_lmo_c2)
seu_lmo_c3$hashedDrops_default_calls <- create_hashedDrops_factor(seu_lmo_c3)HashSolo
HashSolo is a scanpy program. Needs a bit of prep Write to anndata compatible files Counts
library(Matrix)
Attaching package: 'Matrix'The following objects are masked from 'package:tidyr':
expand, pack, unpackThe following object is masked from 'package:S4Vectors':
expandwriteMM(seu_lmo_c1@assays$HTO@counts, here("data", "cell_line_data", "adata", "c1_counts.mtx"))NULLwriteMM(seu_lmo_c2@assays$HTO@counts, here("data", "cell_line_data", "adata", "c2_counts.mtx"))NULLwriteMM(seu_lmo_c3@assays$HTO@counts, here("data", "cell_line_data", "adata", "c3_counts.mtx"))NULLBarcodes
barcodes <- data.frame(colnames(seu_lmo_c1))
colnames(barcodes)<-'Barcode'
write.csv(barcodes, here("data", "cell_line_data", "adata", "c1_barcodes.csv"),
quote = FALSE,row.names = FALSE)
barcodes <- data.frame(colnames(seu_lmo_c2))
colnames(barcodes)<-'Barcode'
write.csv(barcodes, here("data", "cell_line_data", "adata", "c2_barcodes.csv"),
quote = FALSE,row.names = FALSE)
barcodes <- data.frame(colnames(seu_lmo_c3))
colnames(barcodes)<-'Barcode'
write.csv(barcodes, here("data", "cell_line_data", "adata", "c3_barcodes.csv"),
quote = FALSE,row.names = FALSE)Save LMO names (just need one per capture)
HTOs <- data.frame(rownames(seu_lmo_c1))
colnames(HTOs) <- 'HTO'
write.csv(HTOs, here("data", "cell_line_data", "adata", "HTOs.csv"),
quote = FALSE,row.names = FALSE)See hashsolo_calls.ipynb for how we get these assignments
seu_lmo_c1$hashsolo_calls <- read.csv(here("data", "cell_line_data", "adata", "c1_hashsolo.csv"))$Classification
seu_lmo_c2$hashsolo_calls <- read.csv(here("data", "cell_line_data", "adata", "c2_hashsolo.csv"))$Classification
seu_lmo_c3$hashsolo_calls <- read.csv(here("data", "cell_line_data", "adata", "c3_hashsolo.csv"))$ClassificationHTODemux
HDmux <- HTODemux(seu_lmo_c1)Cutoff for CL 01 : 19 readsCutoff for CL 02 : 37 readsCutoff for CL 03 : 45 readsseu_lmo_c1$HTODemux_calls <- HDmux$hash.ID
HDmux <- HTODemux(seu_lmo_c2)Cutoff for CL 01 : 15 readsCutoff for CL 02 : 43 readsCutoff for CL 03 : 50 readsseu_lmo_c2$HTODemux_calls <- HDmux$hash.ID
HDmux <- HTODemux(seu_lmo_c3)Cutoff for CL 01 : 11 readsCutoff for CL 02 : 31 readsCutoff for CL 03 : 38 readsseu_lmo_c3$HTODemux_calls <- HDmux$hash.ID###GMM-Demux
GMM-Demux is run on the command line and needs a function to read in the results and format them all properly.
create_gmm_demux_factor <- function(seu, GMM_path, hto_list) {
#Read in output, have to use the "full" report, not the simplified one.
calls <- read.csv(paste0(GMM_path, "/GMM_full.csv"), row.names = 1)
#Read in names of clusters
cluster_names <- read.table(paste0(GMM_path, "/GMM_full.config"), sep = ",")
names(cluster_names) <- c("Cluster_id", "assignment")
#Need to fix the formatting of the assignment names, for some reason there's a leading space.
cluster_names$assignment <- gsub(x = cluster_names$assignment, pattern = '^ ', replacement = '')
#Add cell barcodes
calls$Barcode <- rownames(calls)
calls <- merge(calls, cluster_names, by = "Cluster_id", sort = FALSE)
#Need to re-order after merge for some reason
calls <- calls[order(match(calls$Barcode, names(seu$Barcode))), ]
#Rename the negative cluster for consistency
calls$assignment[calls$assignment == "negative"] <- "Negative"
#Put all the multiplet states into one assignment category
calls$assignment[!calls$assignment %in% c("Negative", hto_list)] <- "Doublet"
return(as.factor(calls$assignment))
}Need to write transpose of counts matrices to .csv files to run GMM-Demux on command line.
write.csv(t(as.matrix(lmo_counts_c1)), here("data", "cell_line_data", "GMM-Demux", "c1_hto_counts_transpose.csv"))
write.csv(t(as.matrix(lmo_counts_c2)), here("data", "cell_line_data", "GMM-Demux", "c2_hto_counts_transpose.csv"))
write.csv(t(as.matrix(lmo_counts_c3)), here("data", "cell_line_data", "GMM-Demux", "c3_hto_counts_transpose.csv"))See script for running GMM-Demux
Add to objects
seu_lmo_c1$GMMDemux_calls <- create_gmm_demux_factor(seu_lmo_c1, here("data", "cell_line_data", "GMM-Demux", "gmm_out_cell_line_c1", "full_report"), LMO_list)
seu_lmo_c2$GMMDemux_calls <- create_gmm_demux_factor(seu_lmo_c2, here("data", "cell_line_data", "GMM-Demux", "gmm_out_cell_line_c2", "full_report"), LMO_list)
seu_lmo_c3$GMMDemux_calls <- create_gmm_demux_factor(seu_lmo_c3, here("data", "cell_line_data", "GMM-Demux", "gmm_out_cell_line_c3", "full_report"), LMO_list)###deMULTIplex
Next is deMULTIplex, using the Seurat wrapper function MULTIseqDemux for this
seu_lmo_c1$deMULTIplex_calls <- MULTIseqDemux(seu_lmo_c1, autoThresh = TRUE)$MULTI_IDIteration 1Using quantile 0.1Iteration 2Using quantile 0.1seu_lmo_c2$deMULTIplex_calls <- MULTIseqDemux(seu_lmo_c2, autoThresh = TRUE)$MULTI_IDIteration 1
Using quantile 0.1Iteration 2Using quantile 0.1seu_lmo_c3$deMULTIplex_calls <- MULTIseqDemux(seu_lmo_c3, autoThresh = TRUE)$MULTI_IDIteration 1
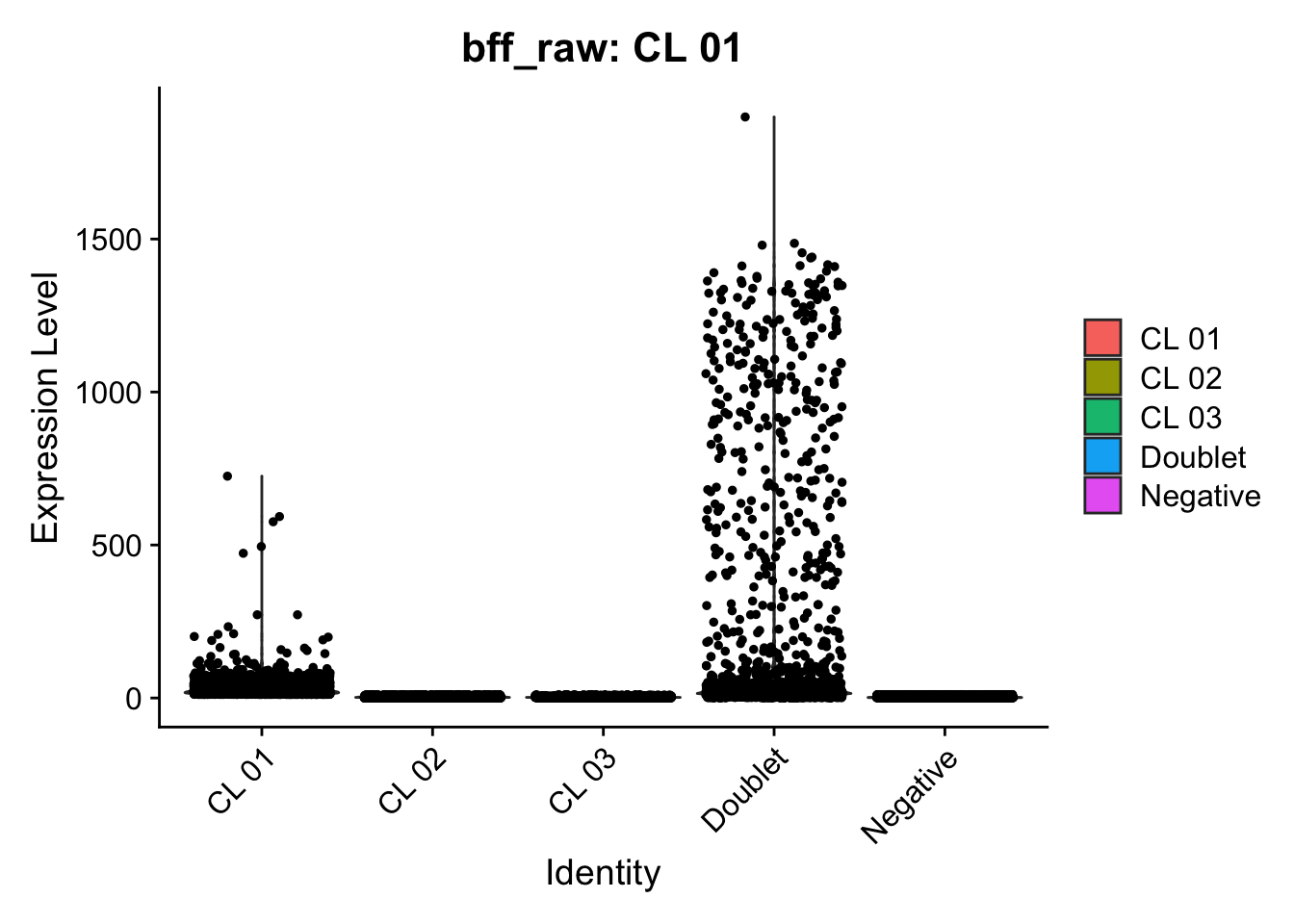
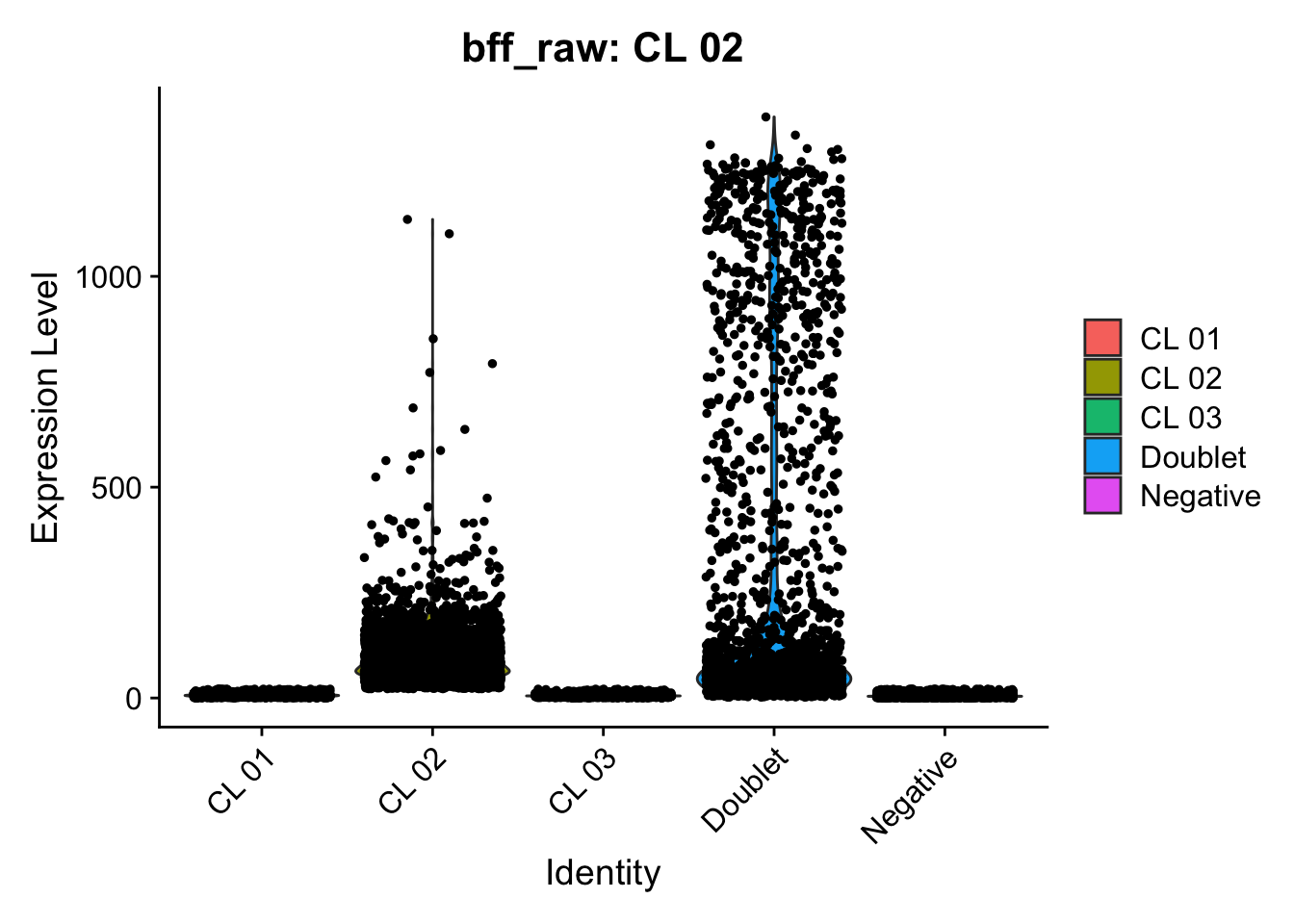
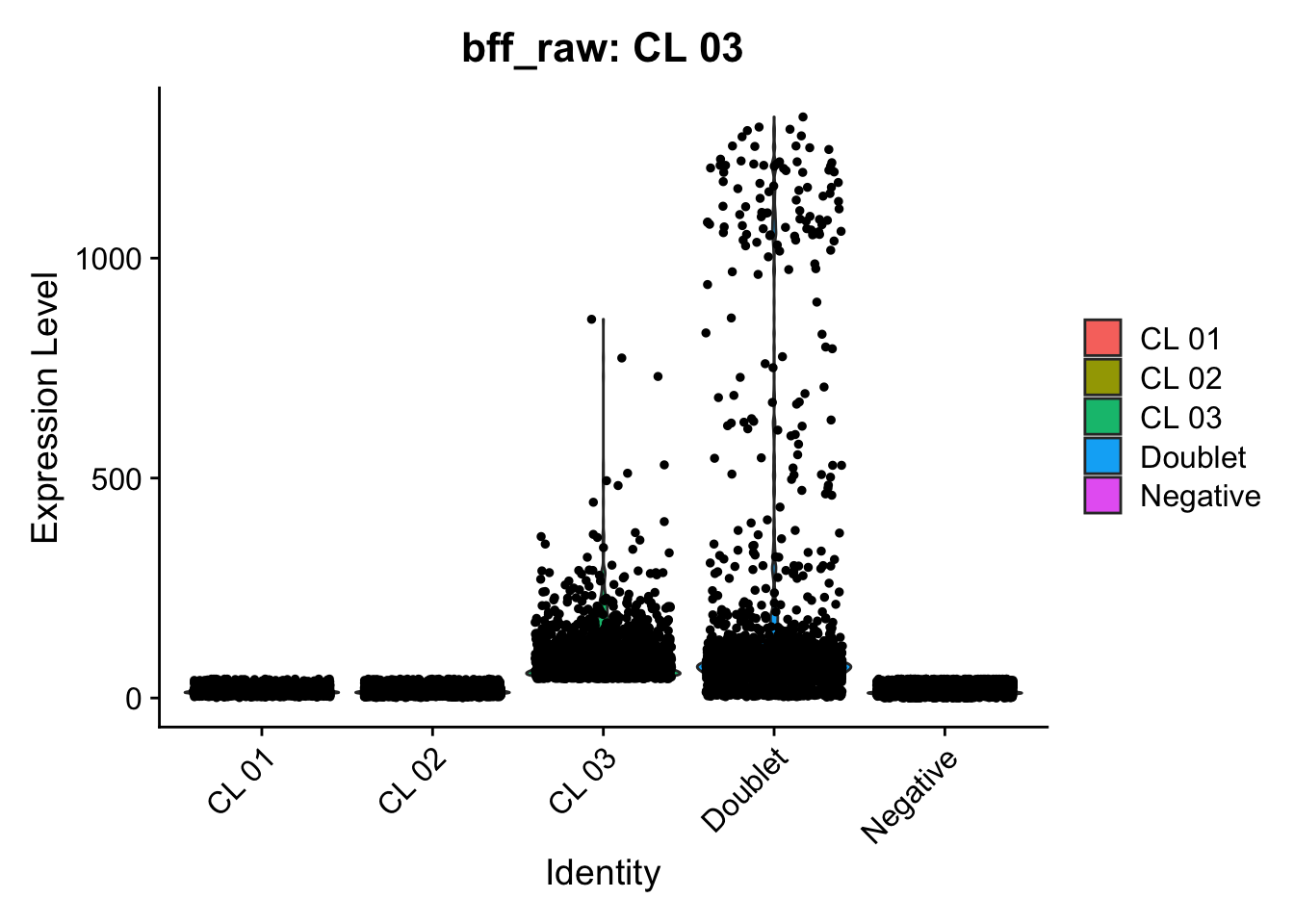
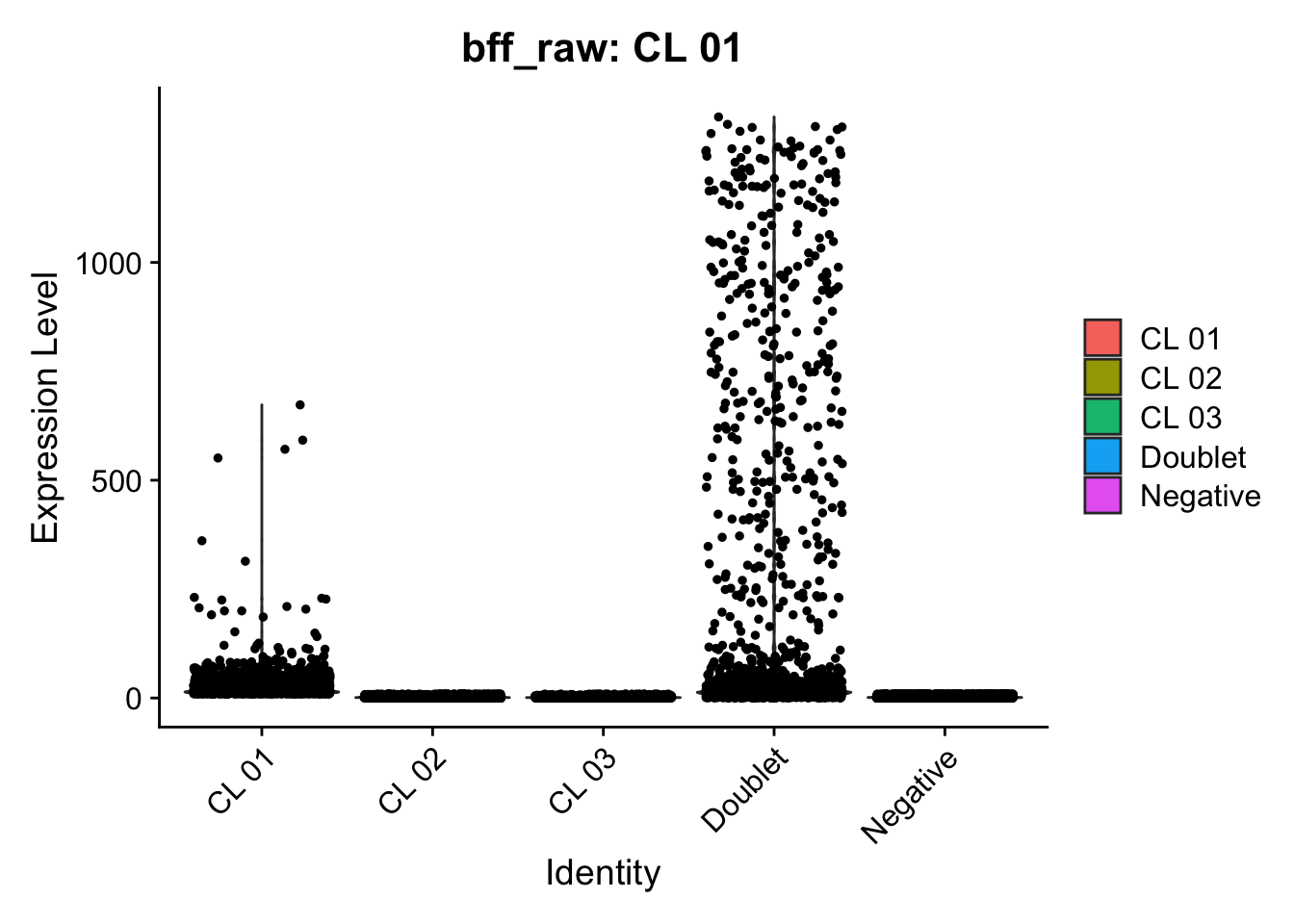
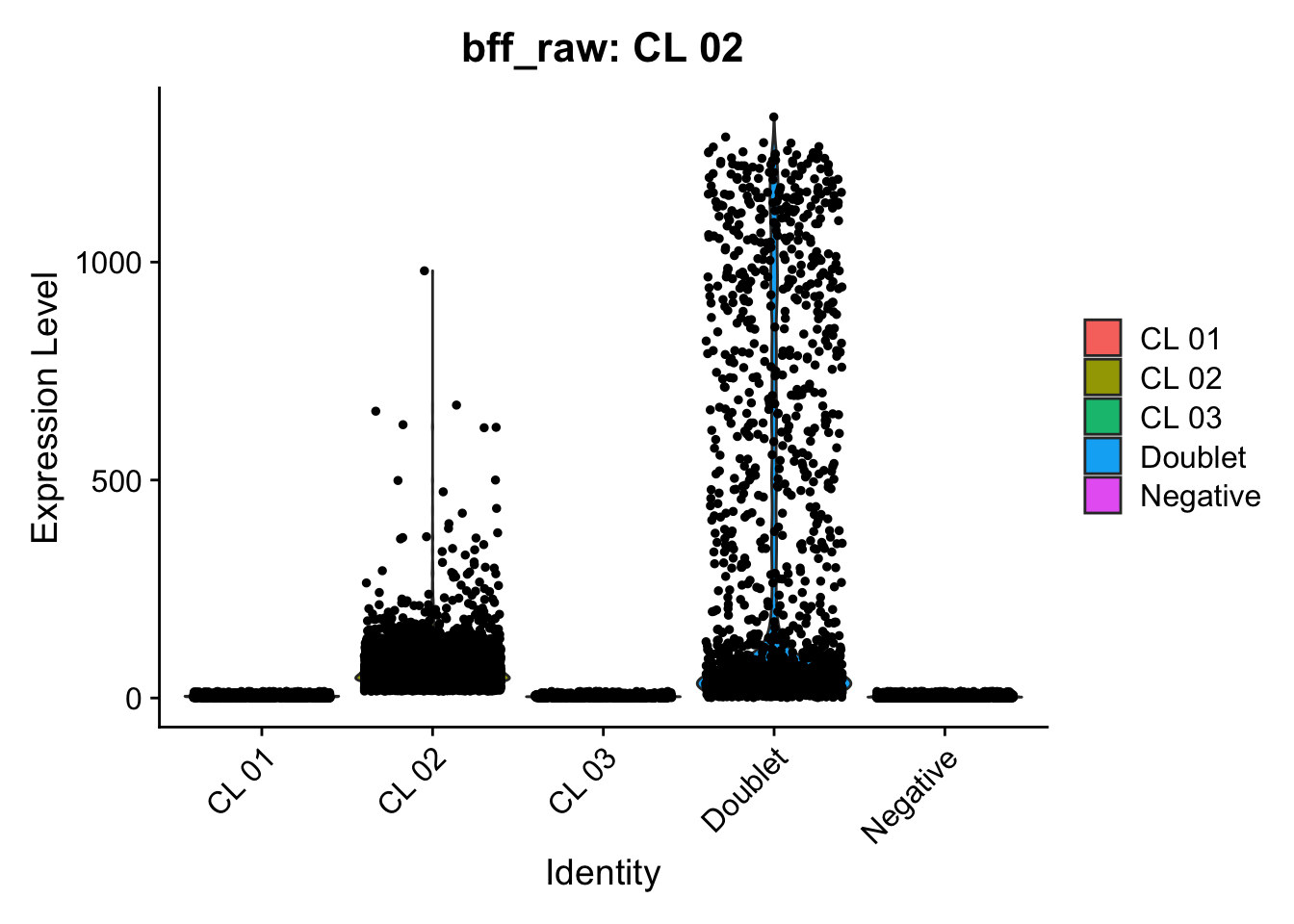
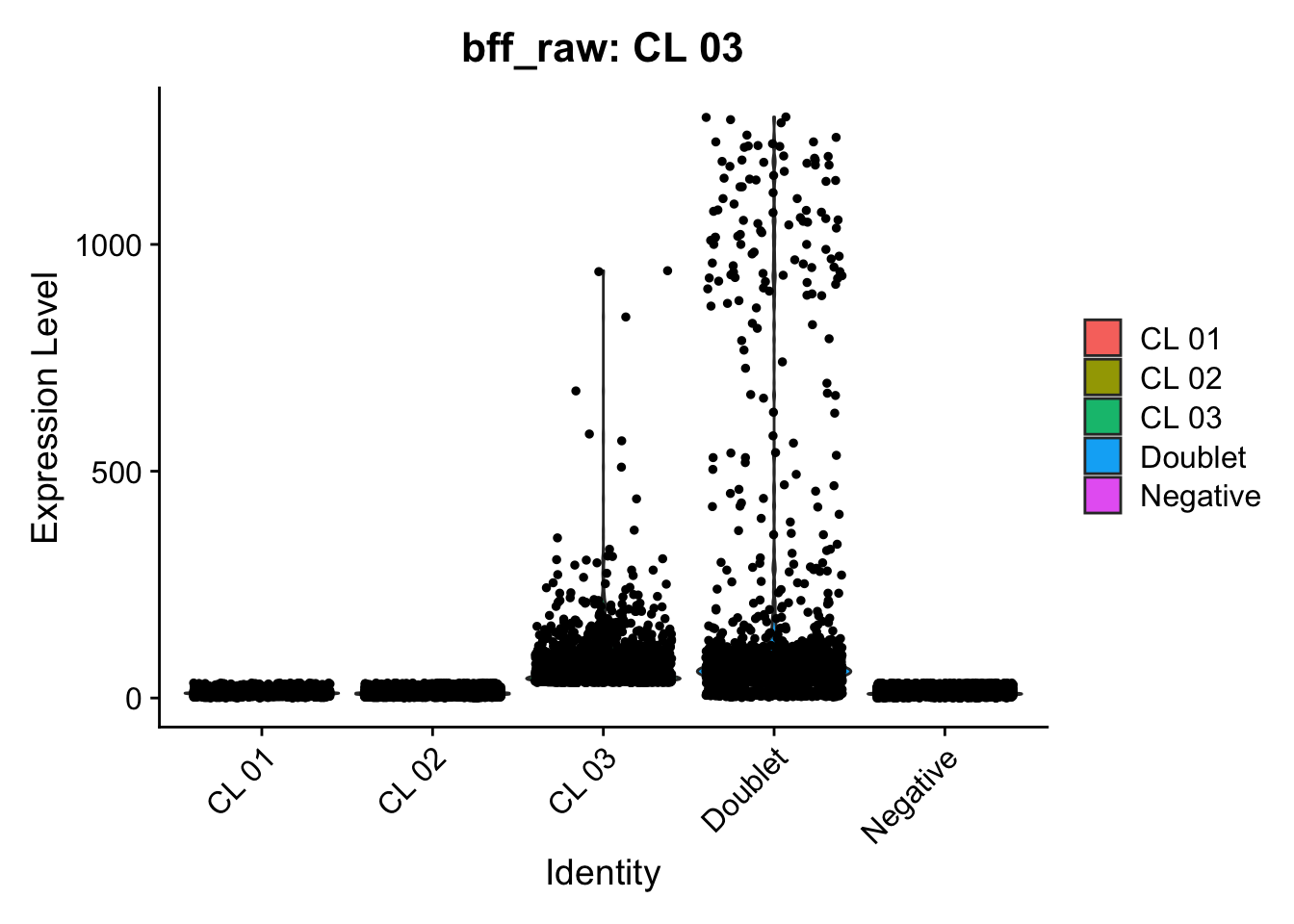
Using quantile 0.1Iteration 2Using quantile 0.1Iteration 3Using quantile 0.1###BFF Finally cellhashR’s BFF_raw and BFF_cluster methods. Need to run this on the raw counts matrix
lmo_counts_c1 <- seu_lmo_c1[["HTO"]]@counts
lmo_counts_c2 <- seu_lmo_c2[["HTO"]]@counts
lmo_counts_c3 <- seu_lmo_c3[["HTO"]]@countscellhashR_calls <- GenerateCellHashingCalls(barcodeMatrix = lmo_counts_c1, methods = c("bff_raw", "bff_cluster"), doTSNE = FALSE, doHeatmap = FALSE)[1] "Starting BFF"
[1] "rows dropped for low counts: 0 of 3"
[1] "Running BFF_raw"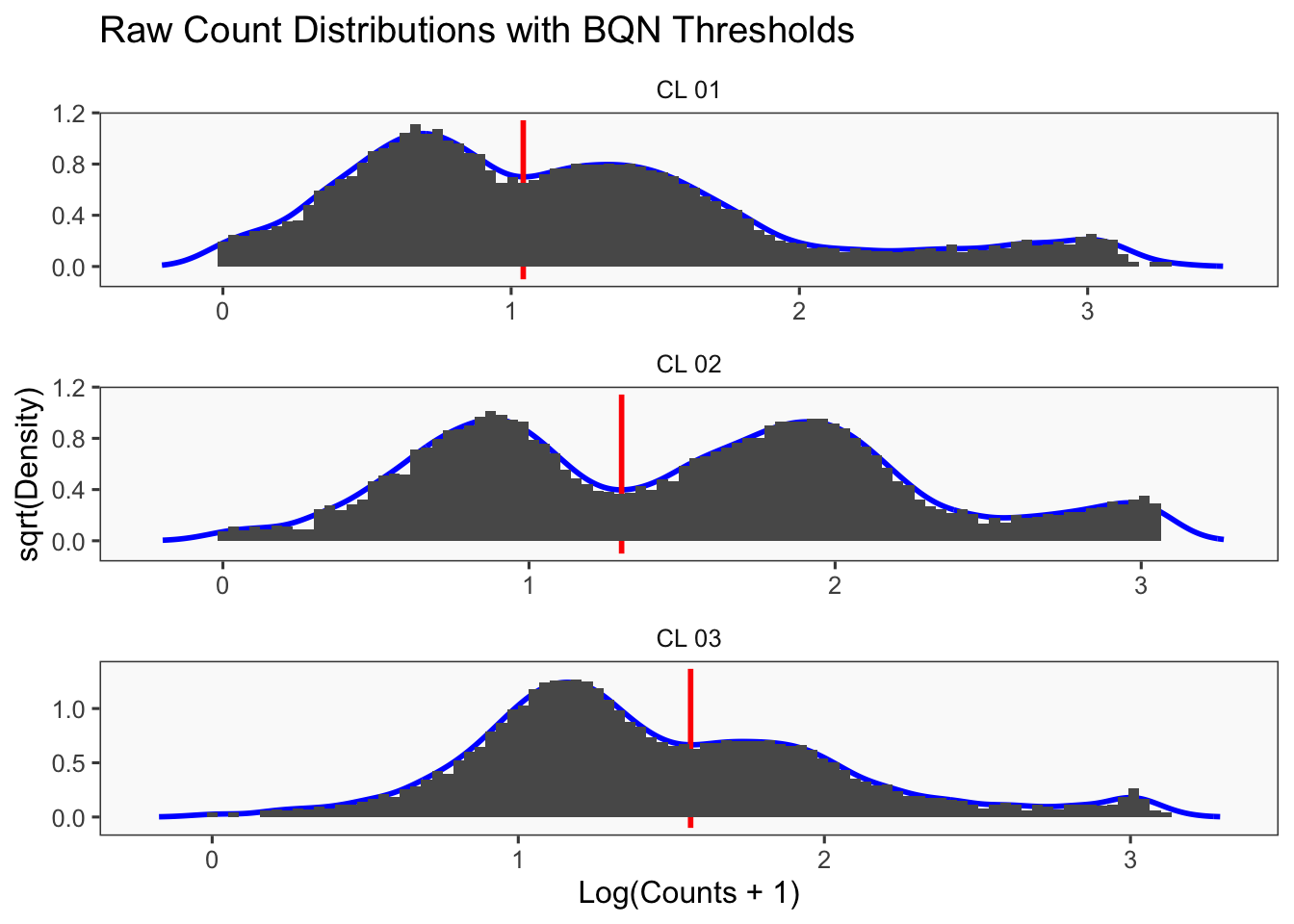
[1] "Thresholds:"
[1] "CL 03: 36.560314666559"
[1] "CL 02: 20.0685207451422"
[1] "CL 01: 11.0152777585693"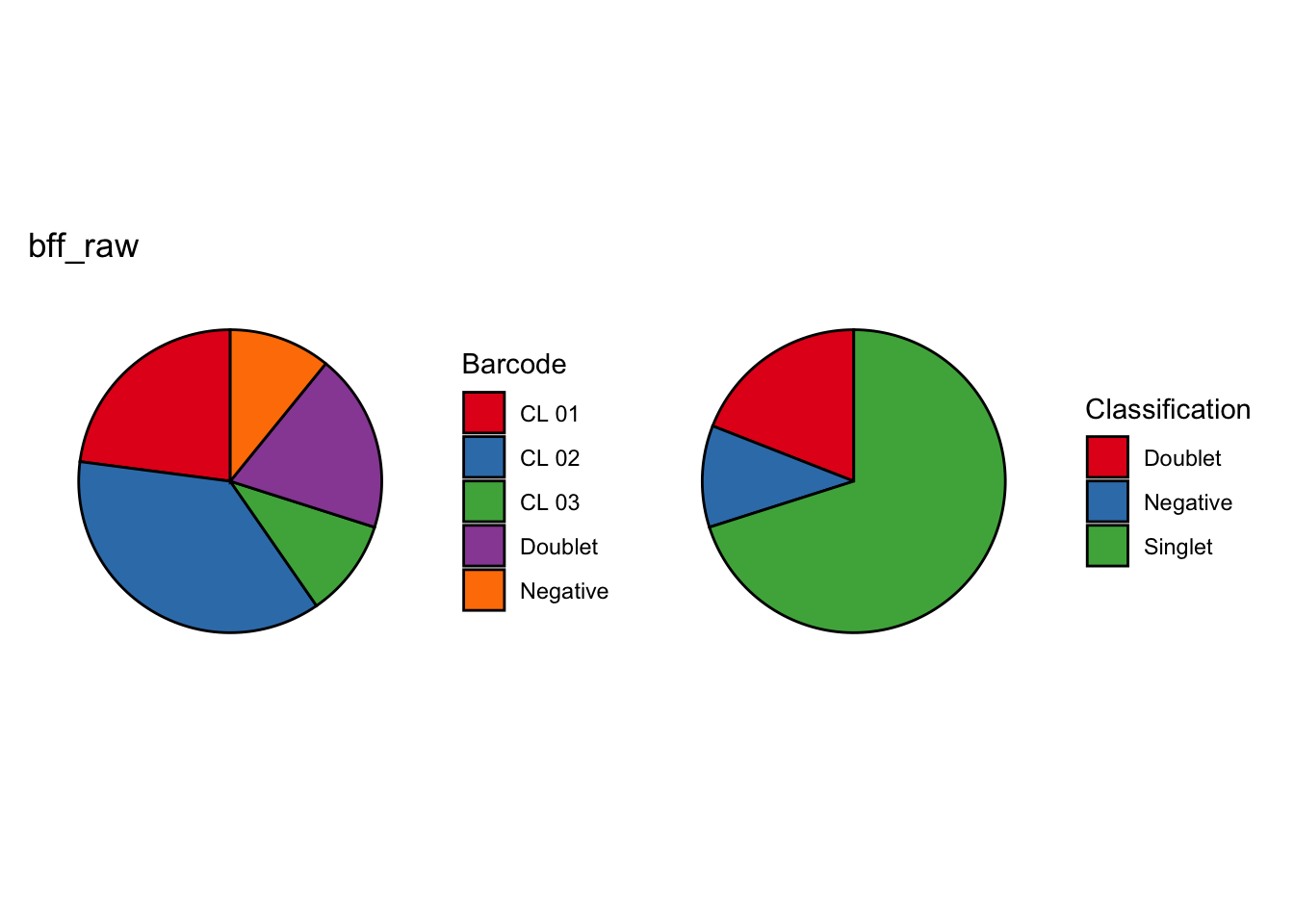
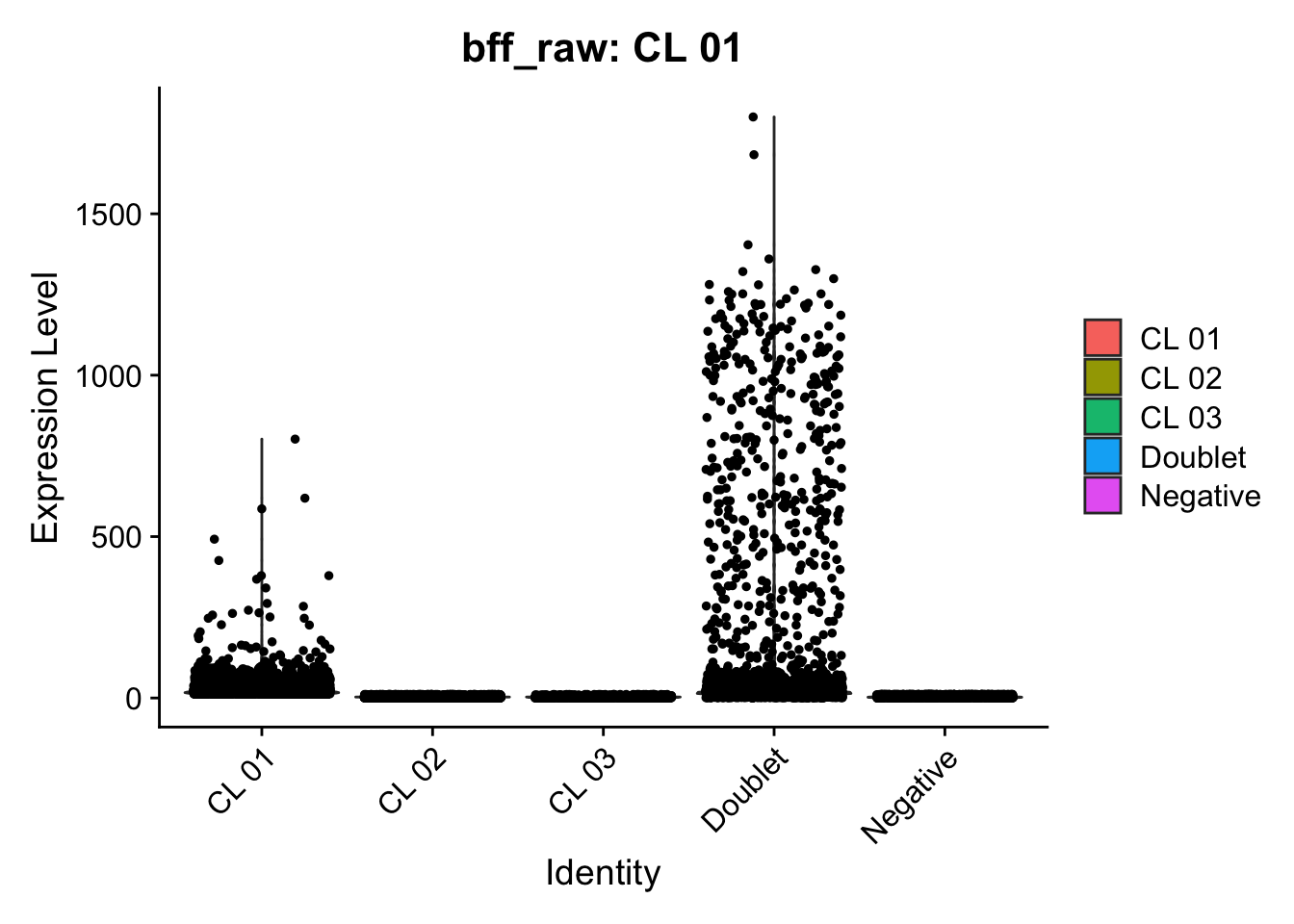
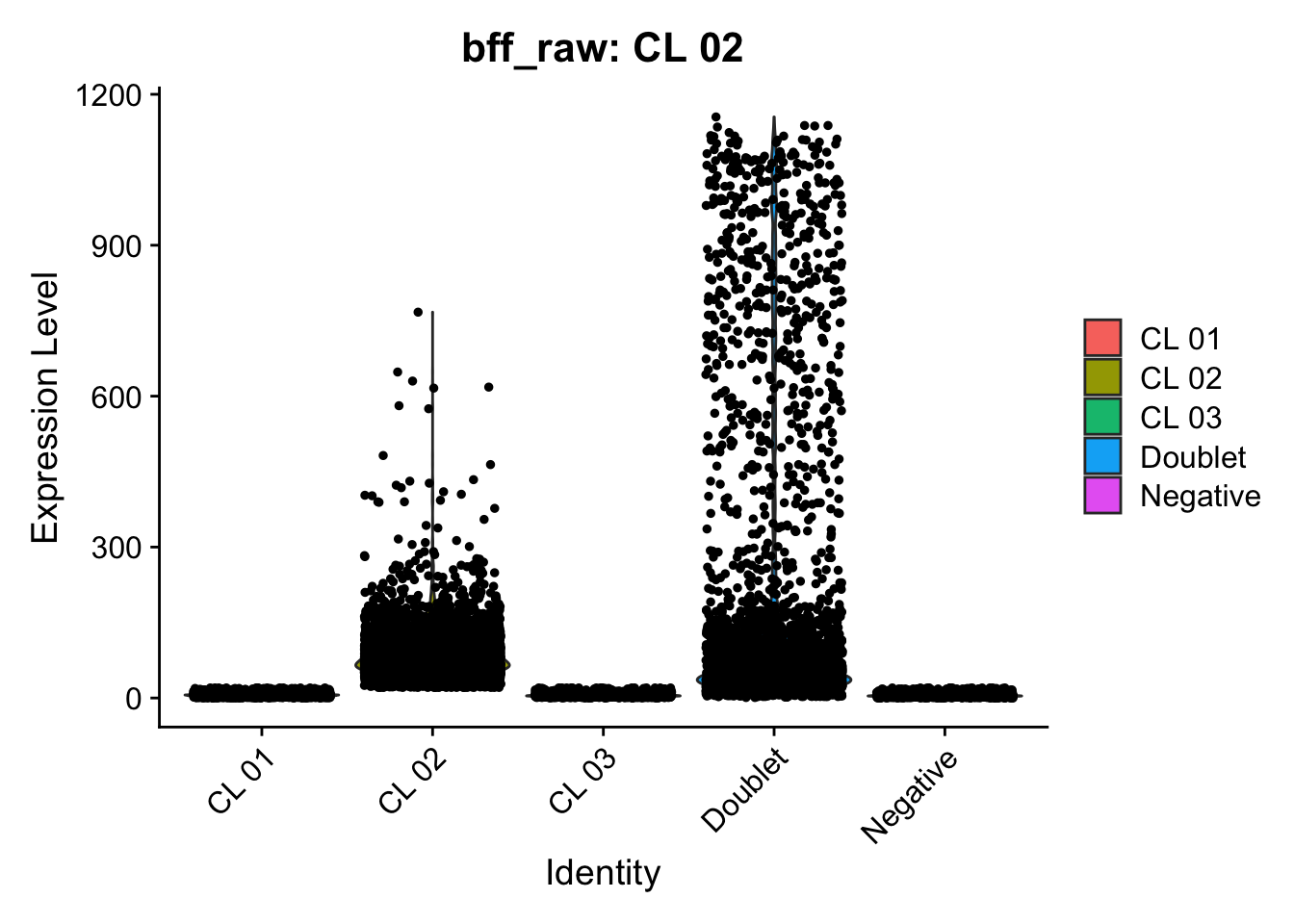
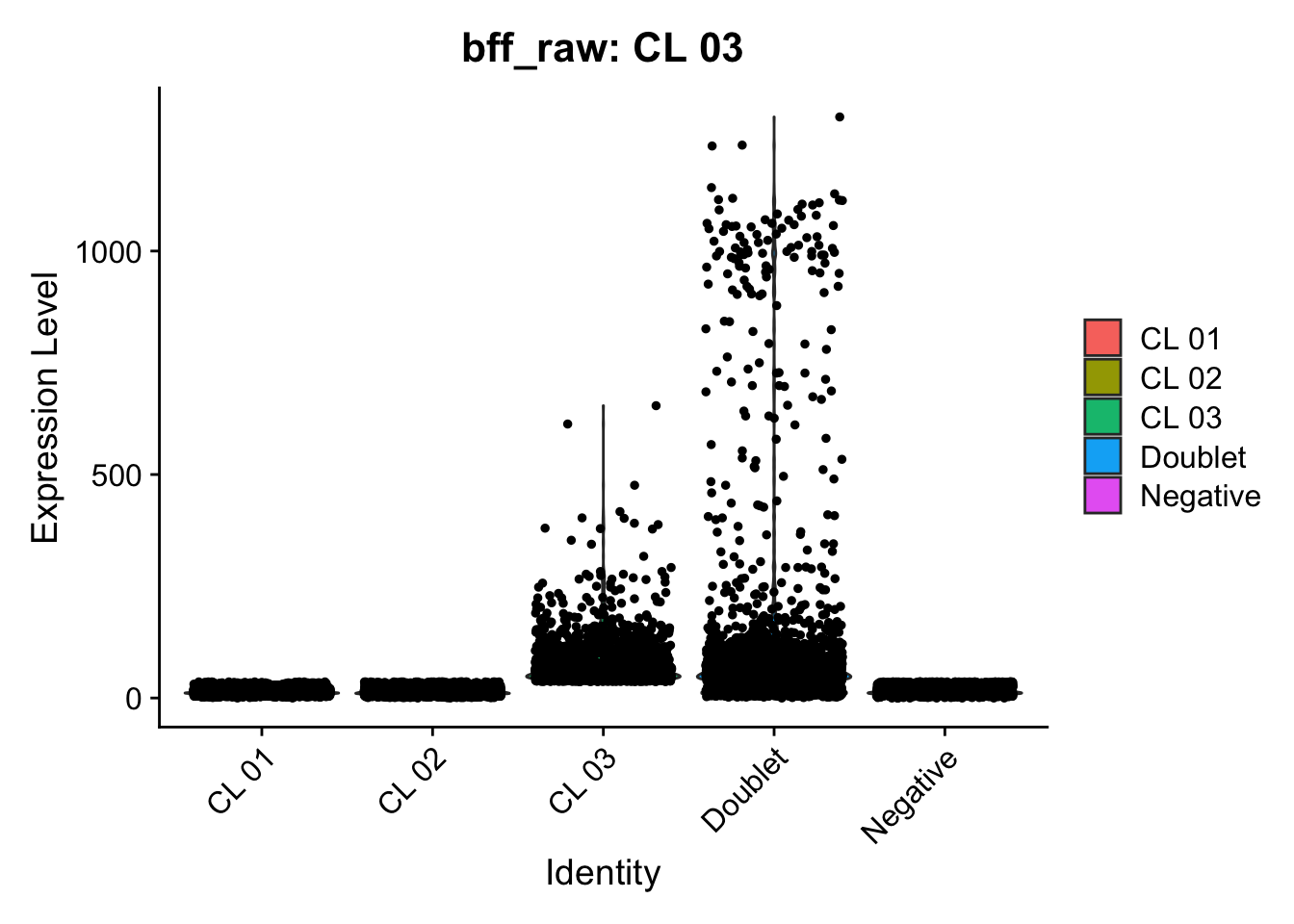
[1] "Starting BFF"
[1] "rows dropped for low counts: 0 of 3"
[1] "Running BFF_cluster"
[1] "Doublet threshold: 0.05"
[1] "Neg threshold: 0.05"
[1] "Min distance as fraction of distance between peaks: 0.1"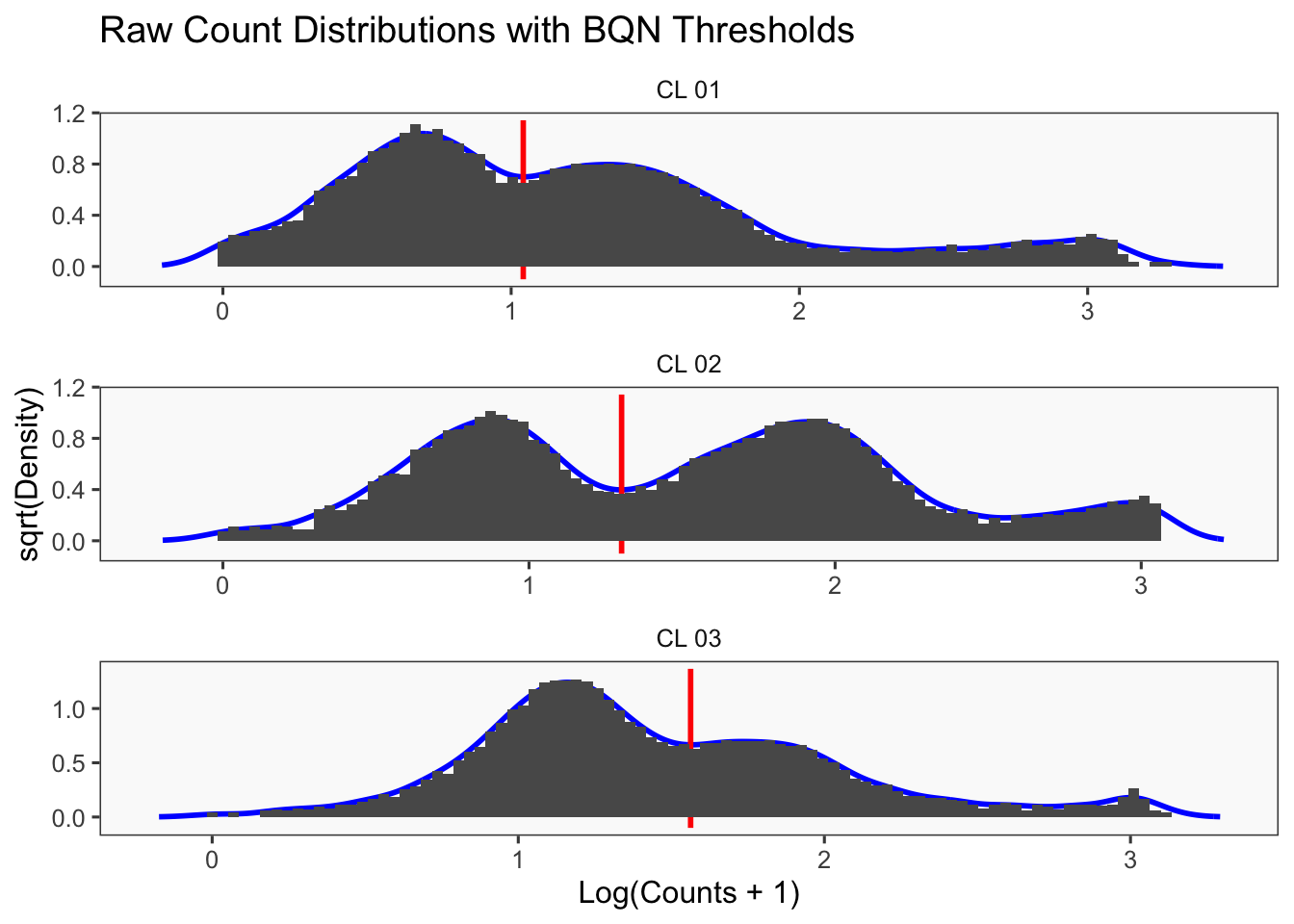
[1] "Thresholds:"
[1] "CL 03: 36.560314666559"
[1] "CL 02: 20.0685207451422"
[1] "CL 01: 11.0152777585693"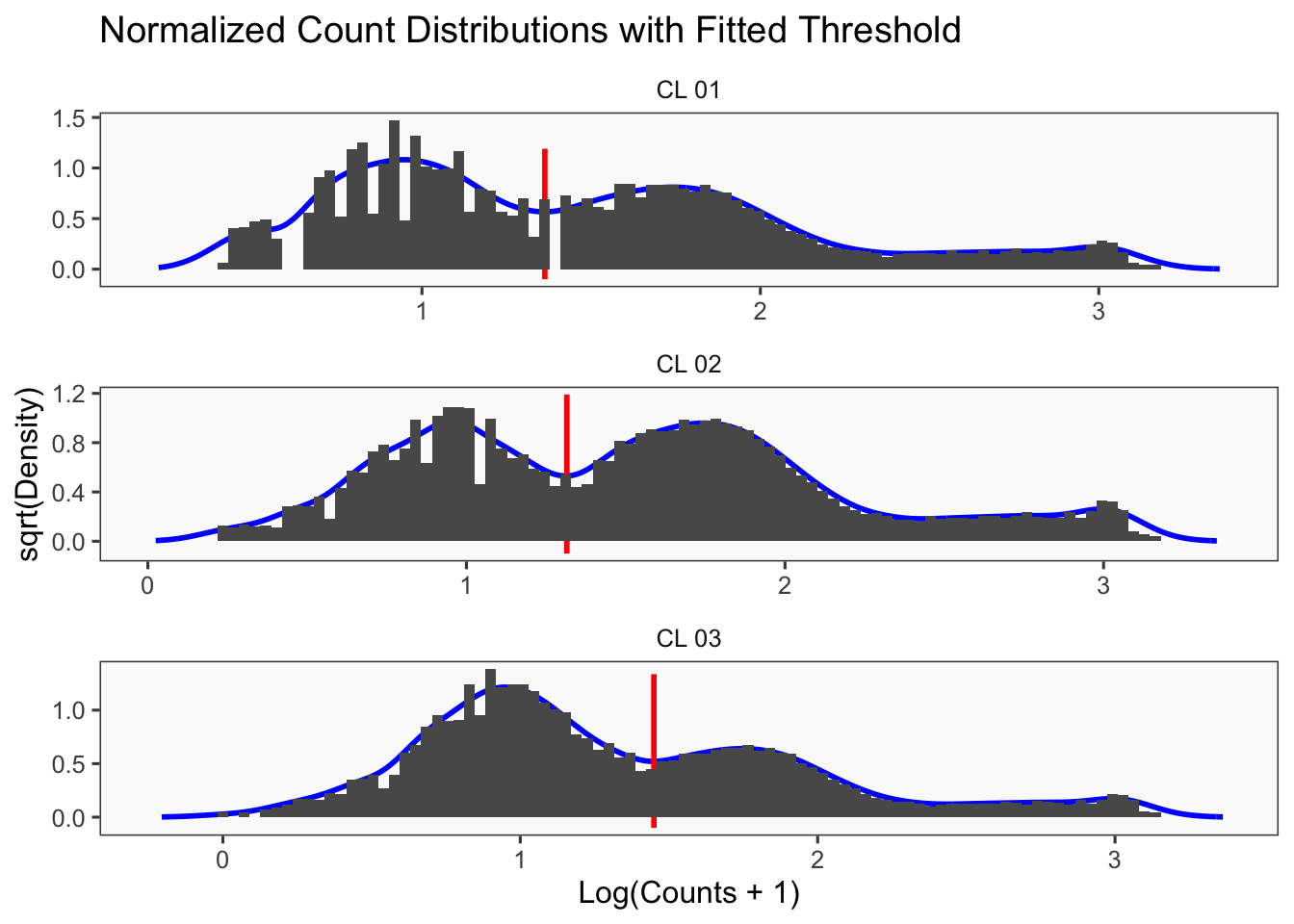
[1] "Smoothing parameter j = 5"
[1] "Smoothing parameter j = 10"
[1] "Smoothing parameter j = 15"
[1] "Smoothing parameter j = 20"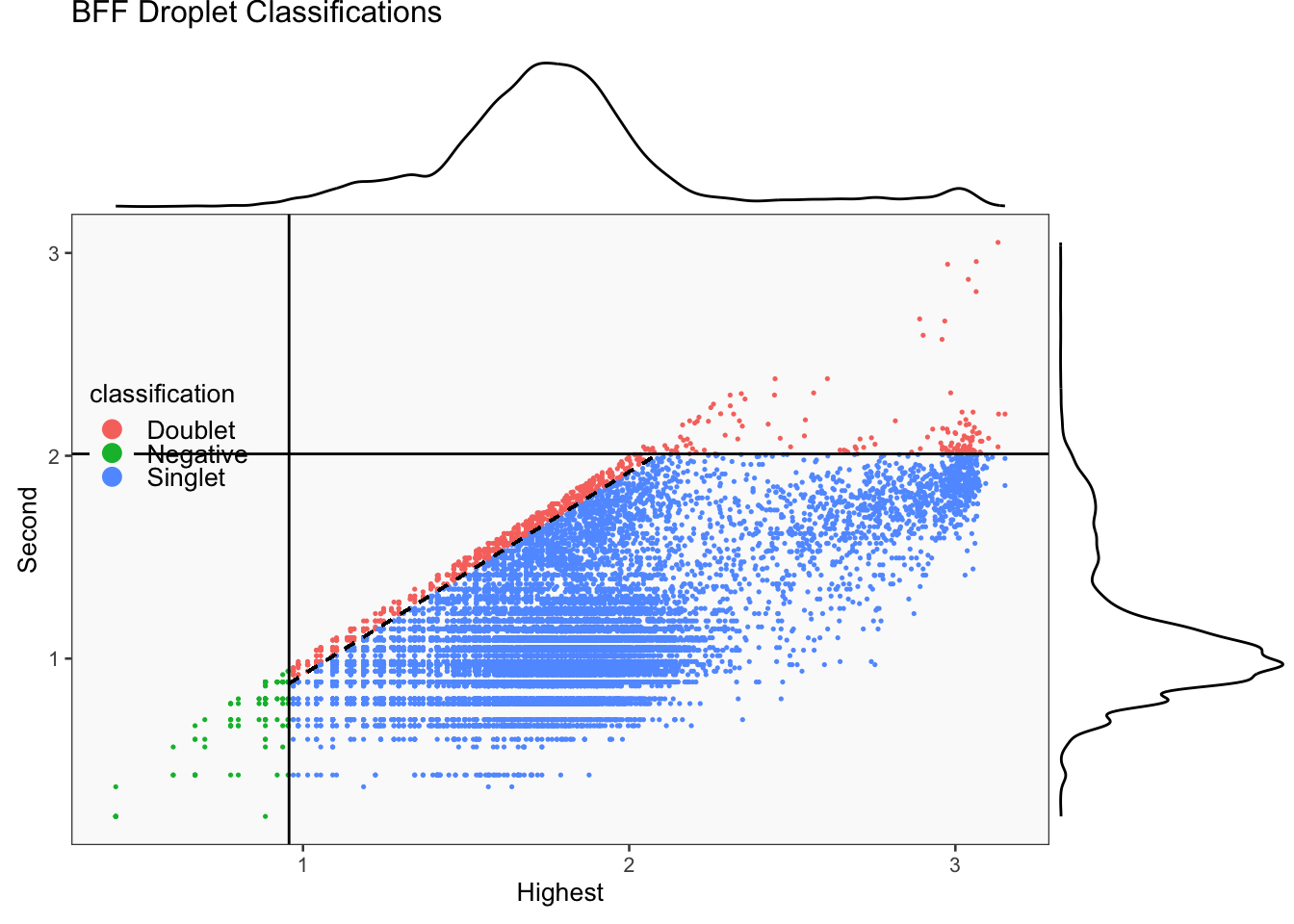
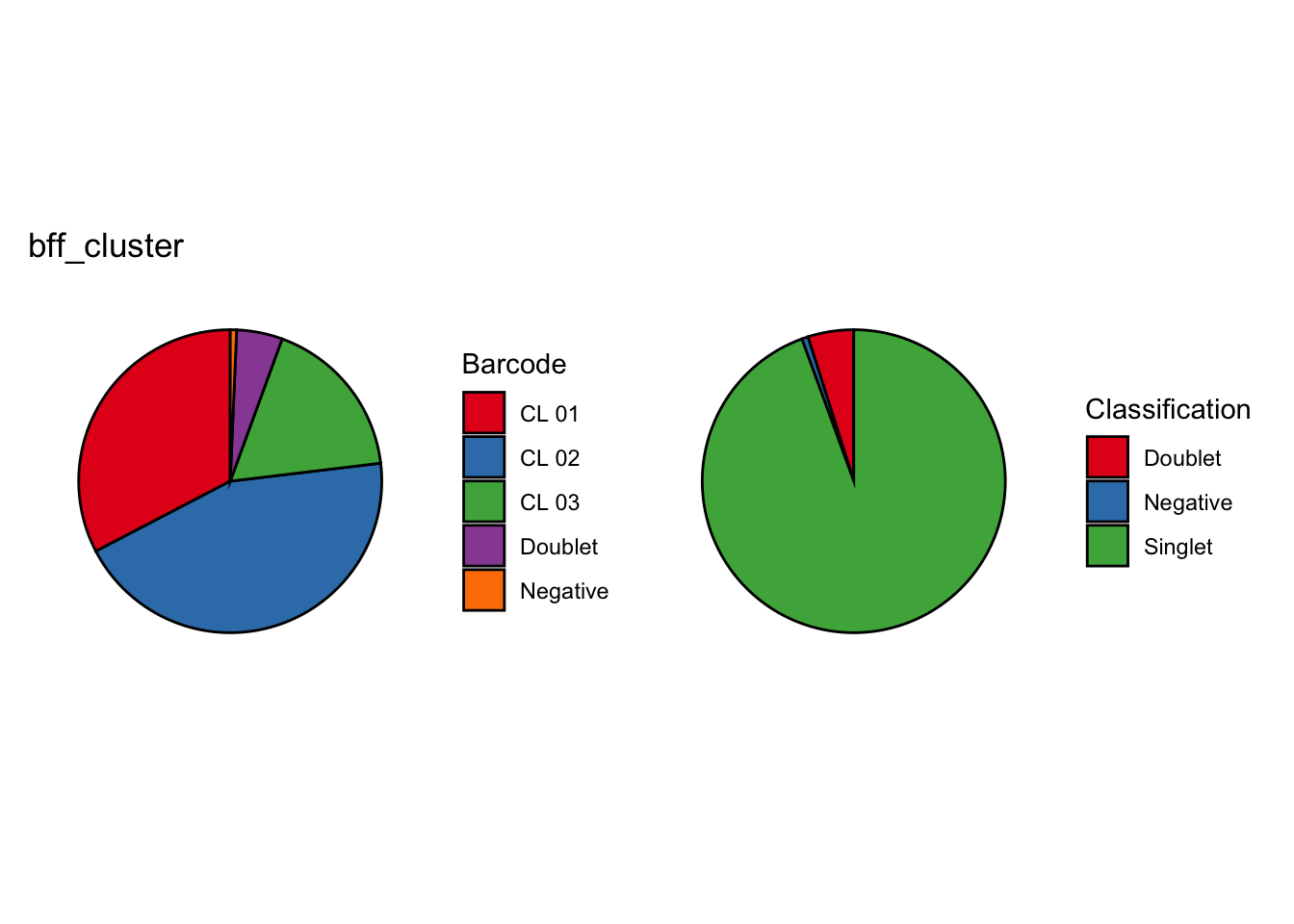
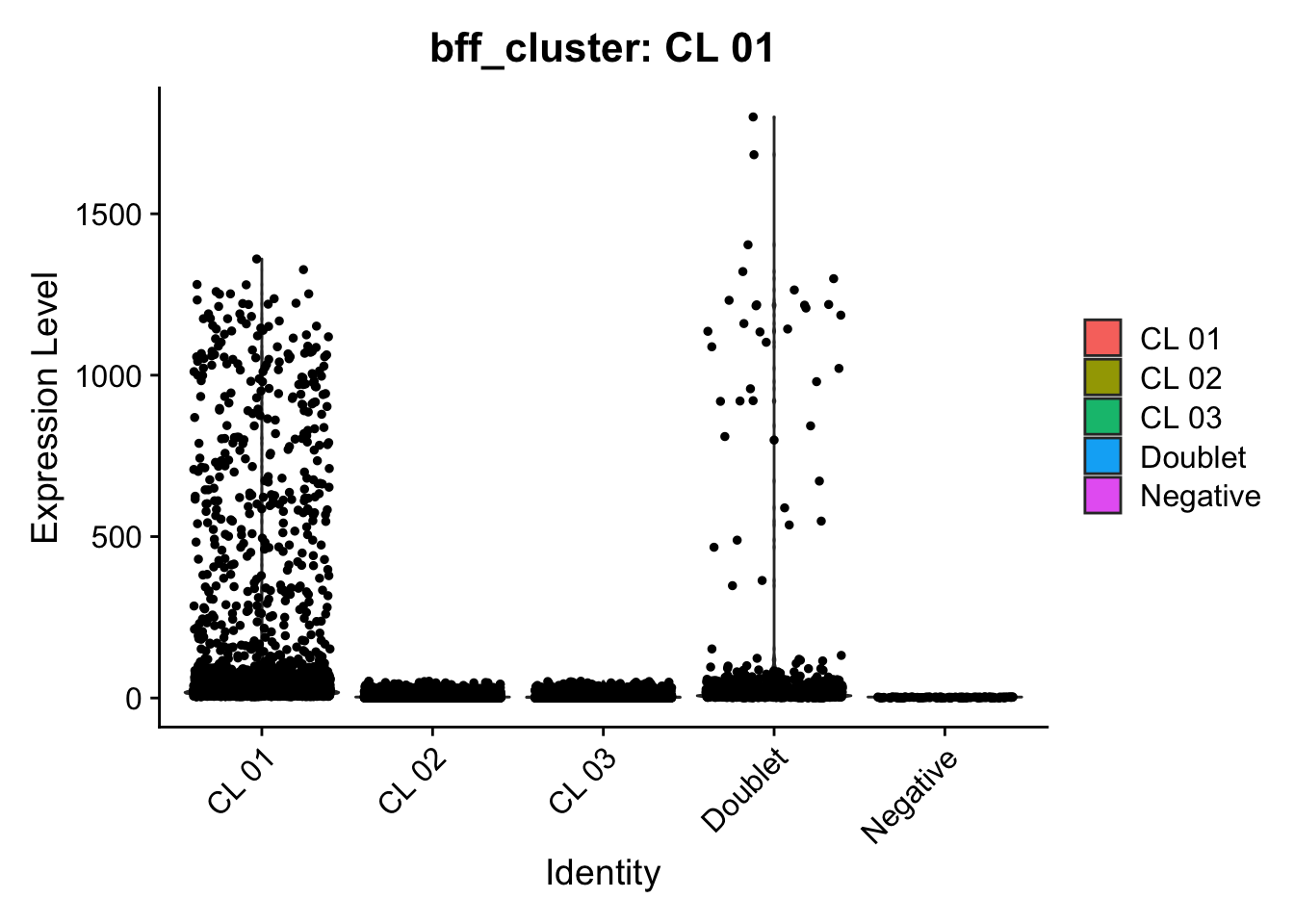
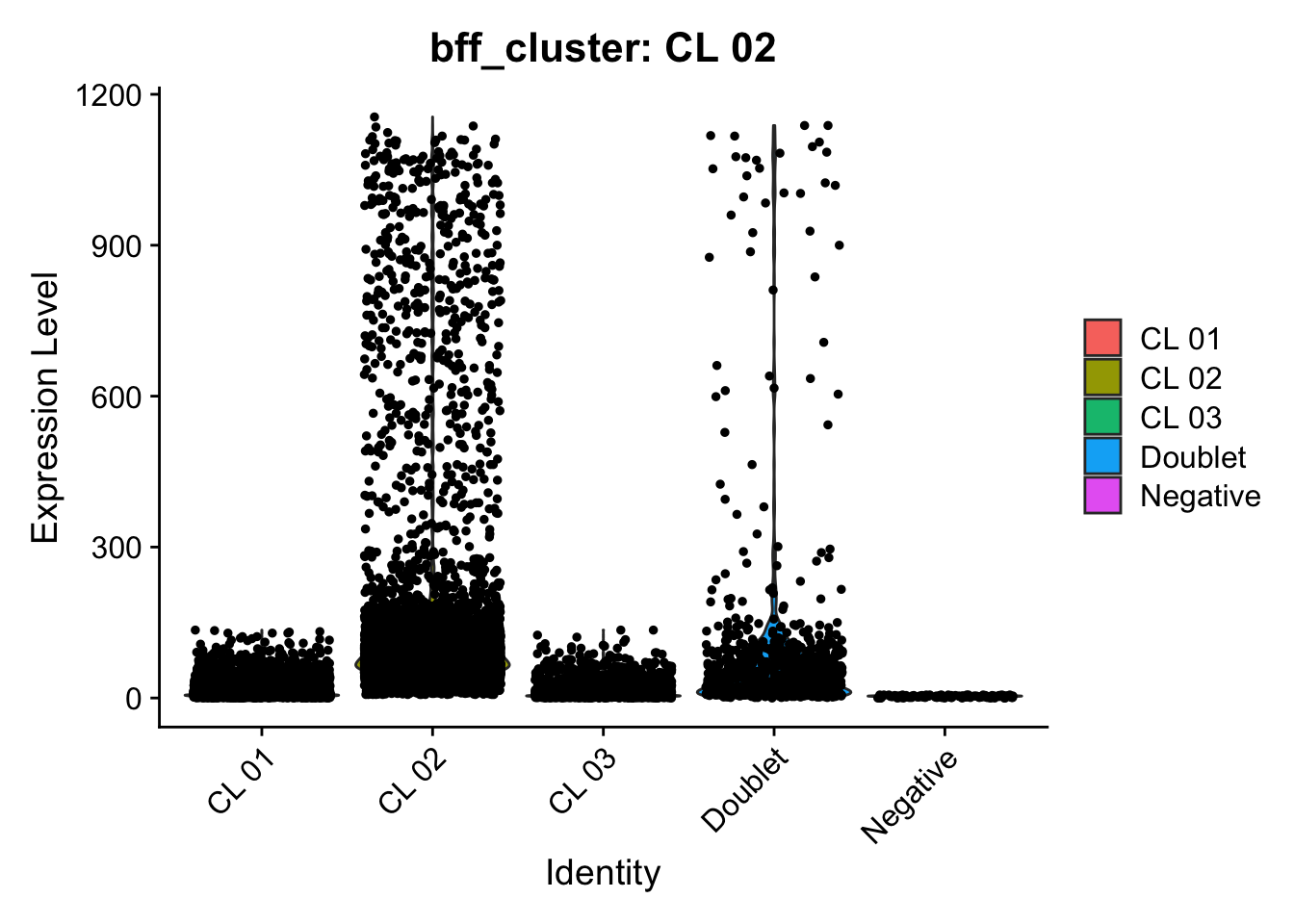
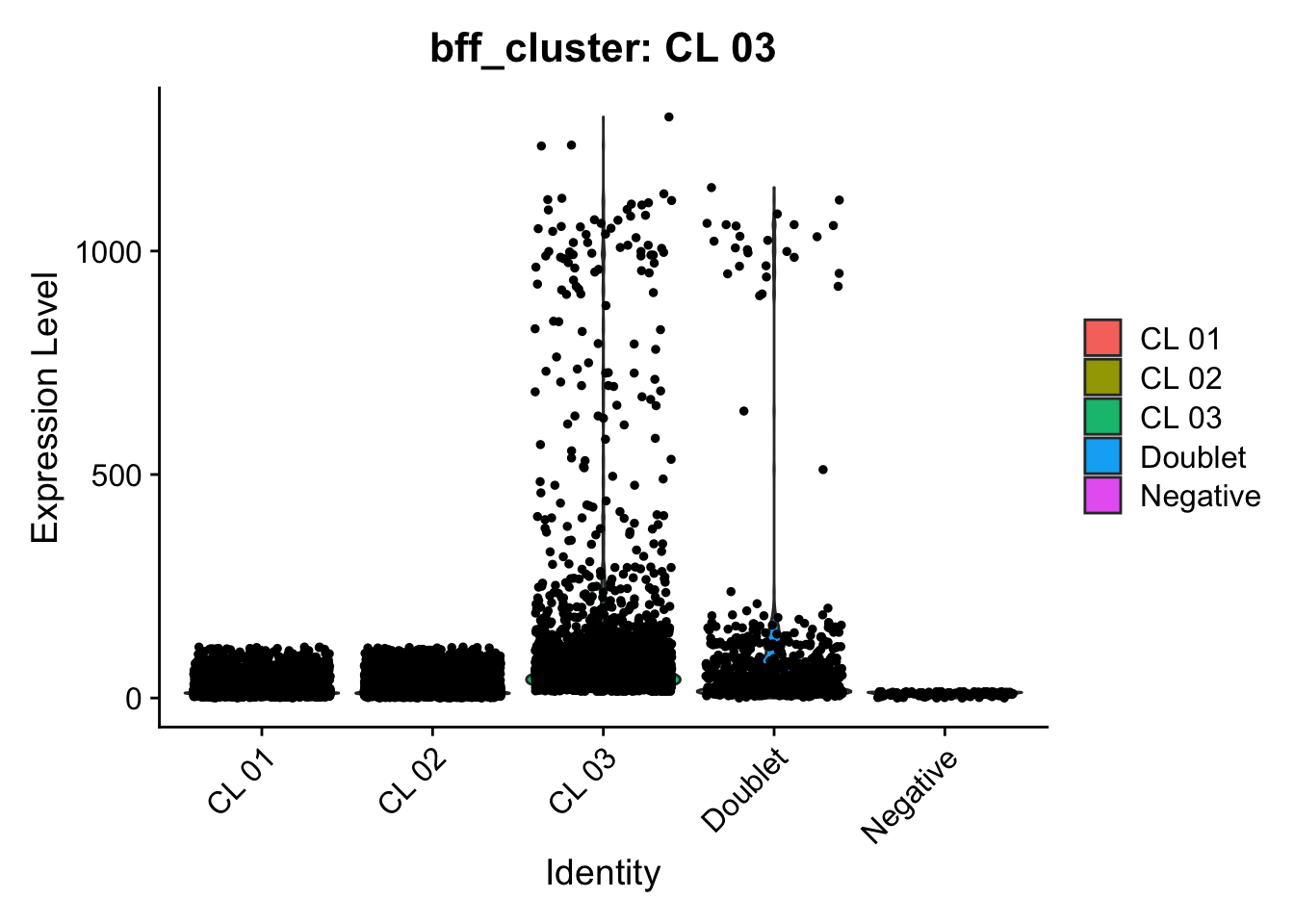
[1] "Generating consensus calls"
[1] "Consensus calls will be generated using: bff_raw,bff_cluster"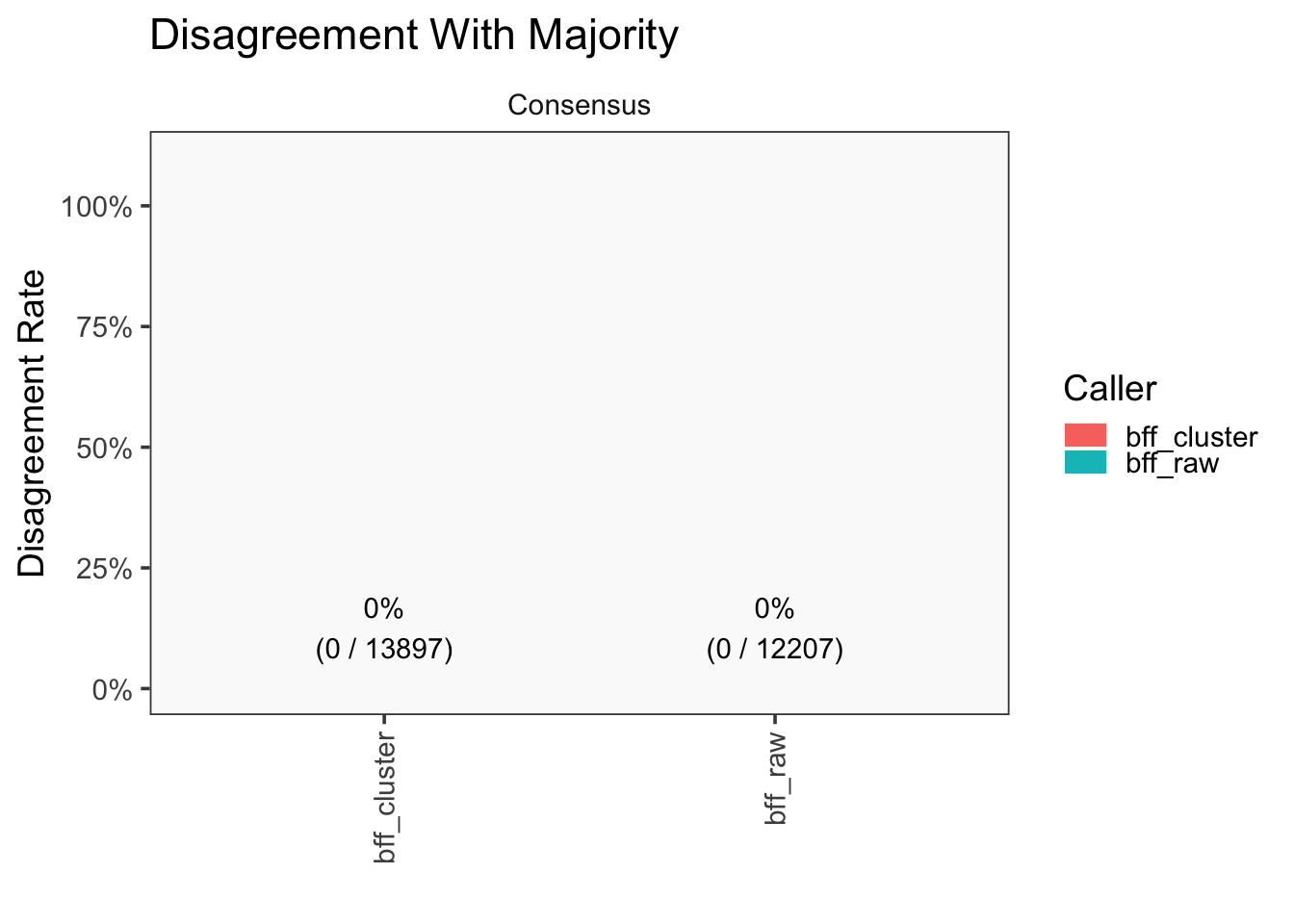
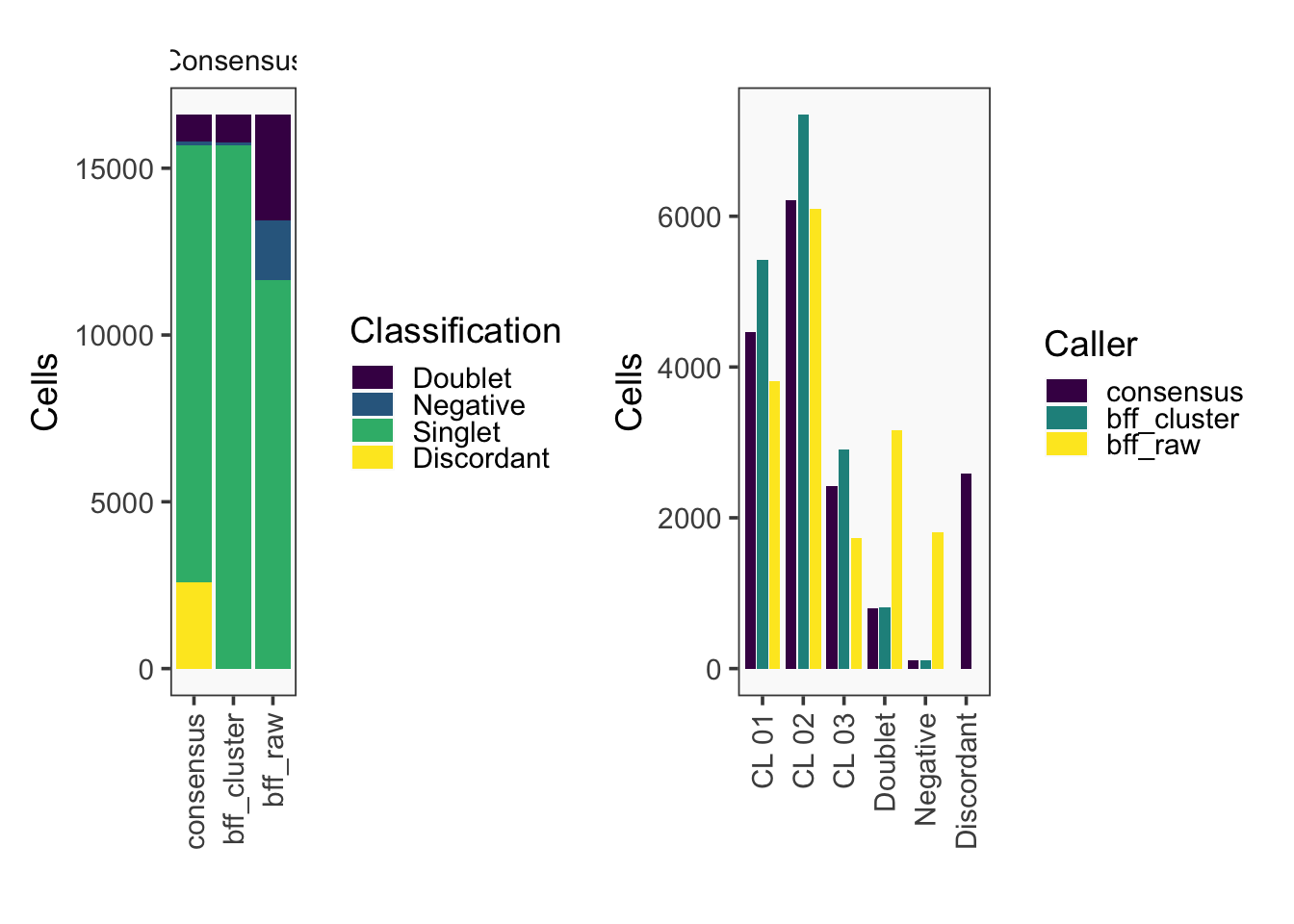
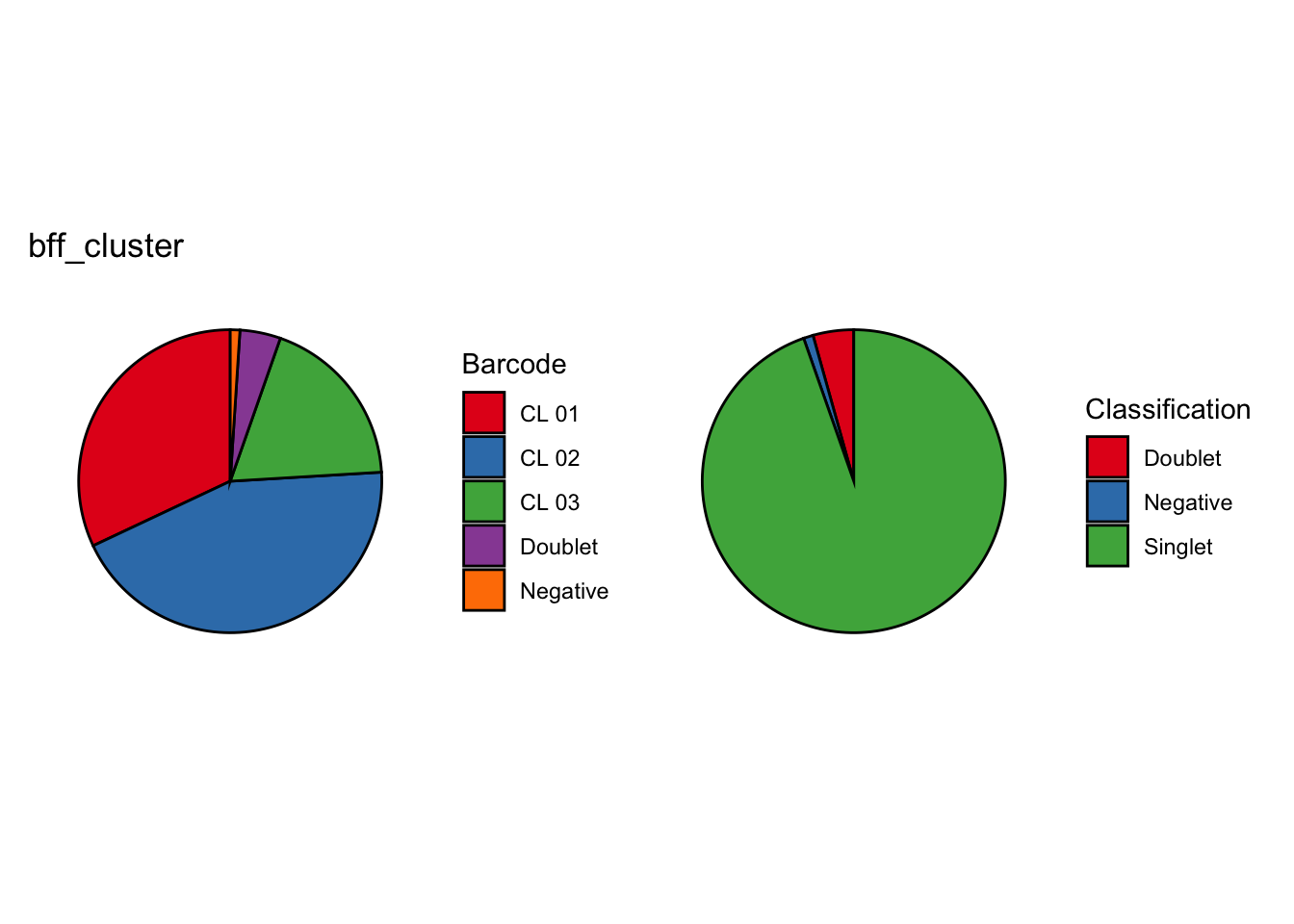
[1] "Total concordant: 14009"
[1] "Total discordant: 2589 (15.6%)"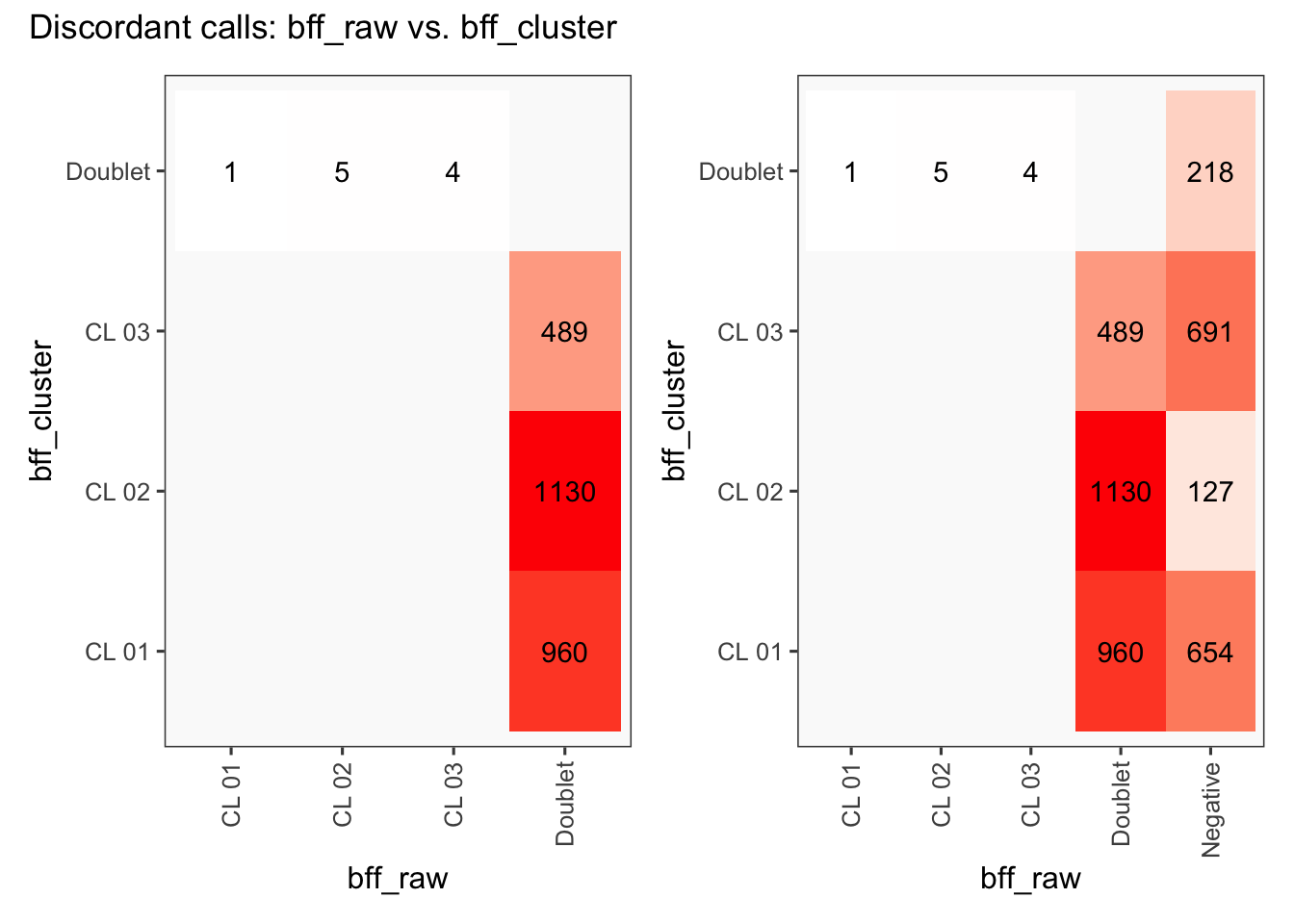
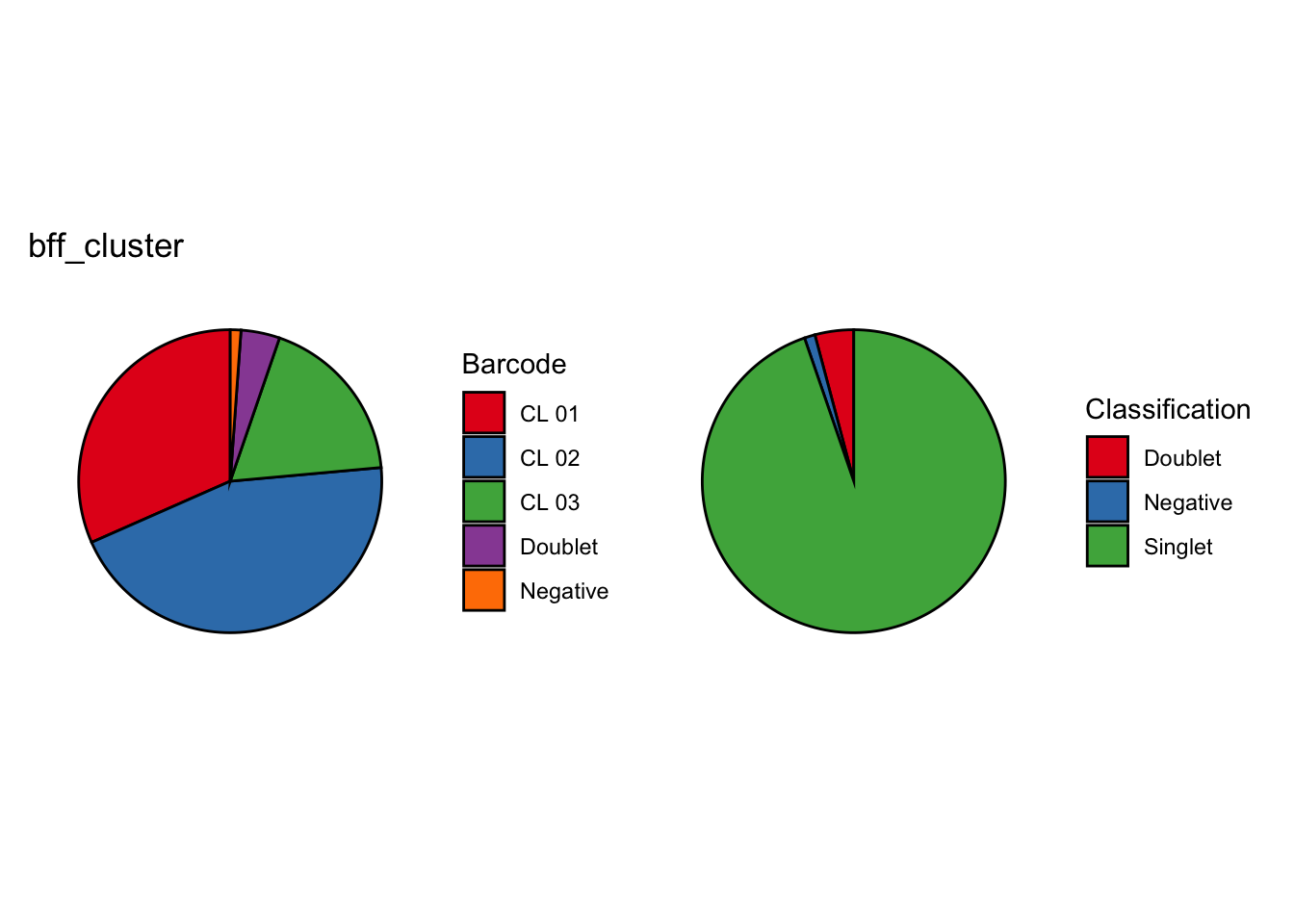
seu_lmo_c1$BFF_raw_calls <- cellhashR_calls$bff_raw
seu_lmo_c1$BFF_cluster_calls <- cellhashR_calls$bff_cluster
cellhashR_calls <- GenerateCellHashingCalls(barcodeMatrix = lmo_counts_c2, methods = c("bff_raw", "bff_cluster"), doTSNE = FALSE, doHeatmap = FALSE)[1] "Starting BFF"
[1] "rows dropped for low counts: 0 of 3"
[1] "Running BFF_raw"
[1] "Thresholds:"
[1] "CL 03: 43.6531370779815"
[1] "CL 02: 21.2184837652457"
[1] "CL 01: 11.3133894314056"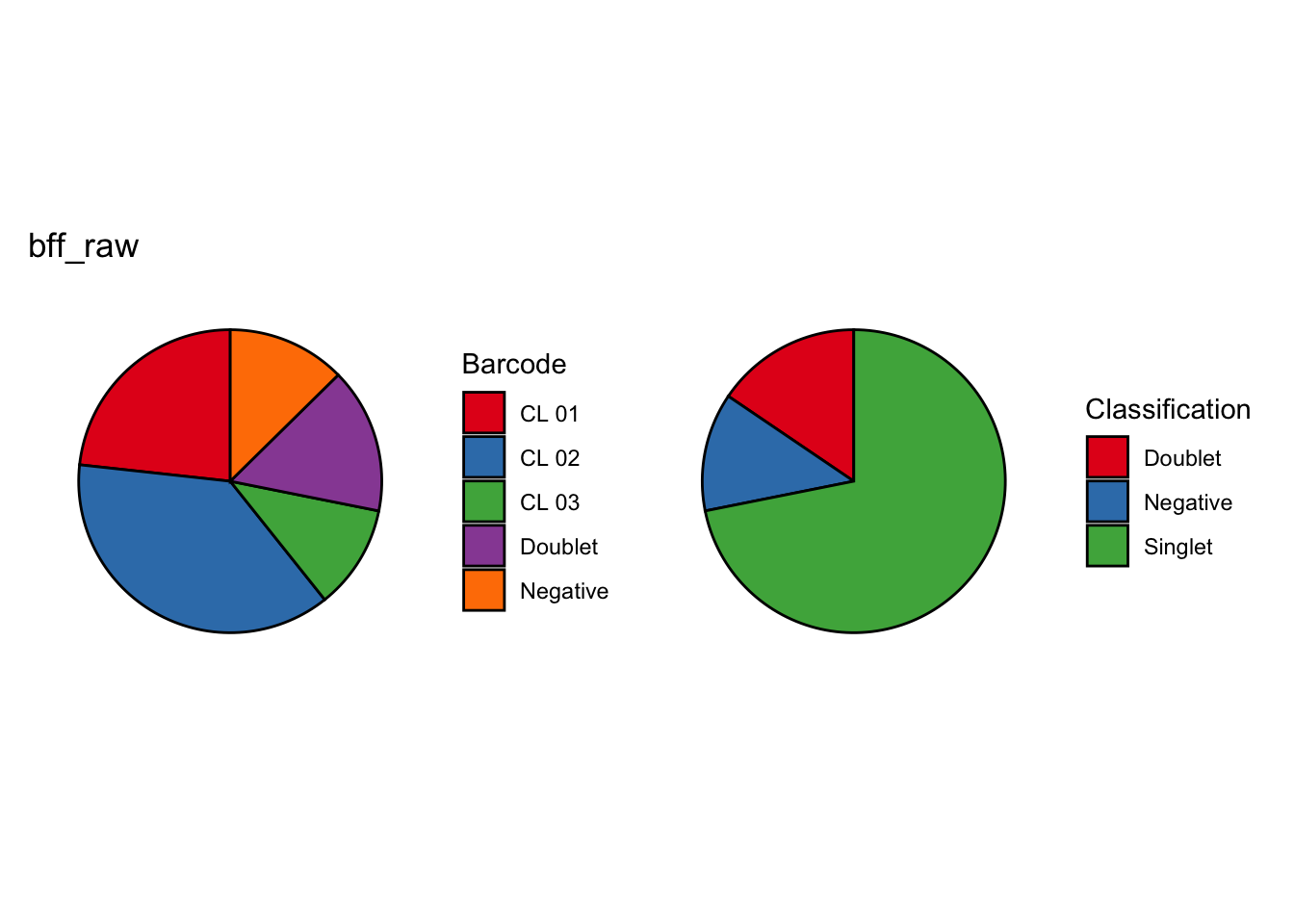
[1] "Starting BFF"
[1] "rows dropped for low counts: 0 of 3"
[1] "Running BFF_cluster"
[1] "Doublet threshold: 0.05"
[1] "Neg threshold: 0.05"
[1] "Min distance as fraction of distance between peaks: 0.1"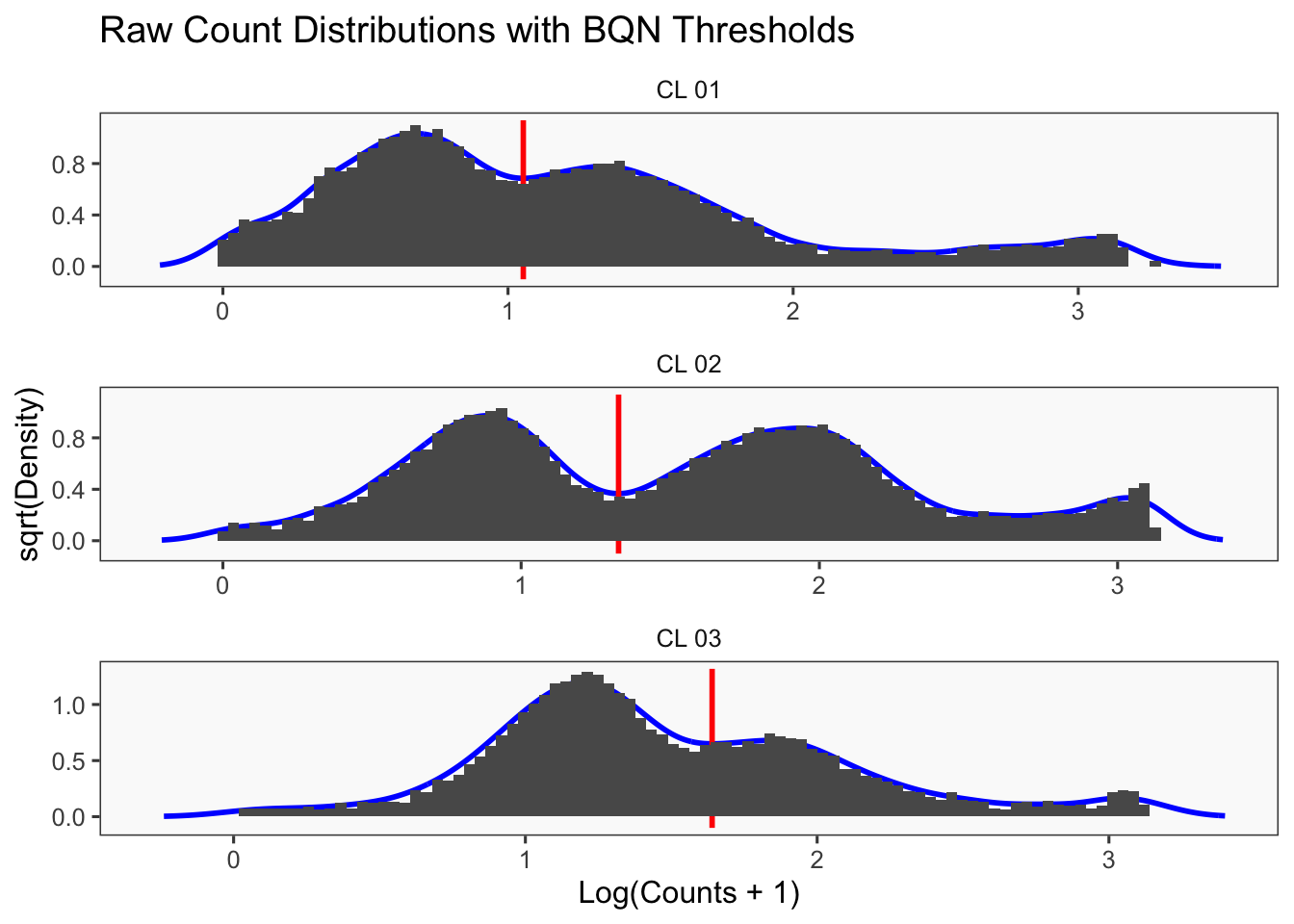
[1] "Thresholds:"
[1] "CL 03: 43.6531370779815"
[1] "CL 02: 21.2184837652457"
[1] "CL 01: 11.3133894314056"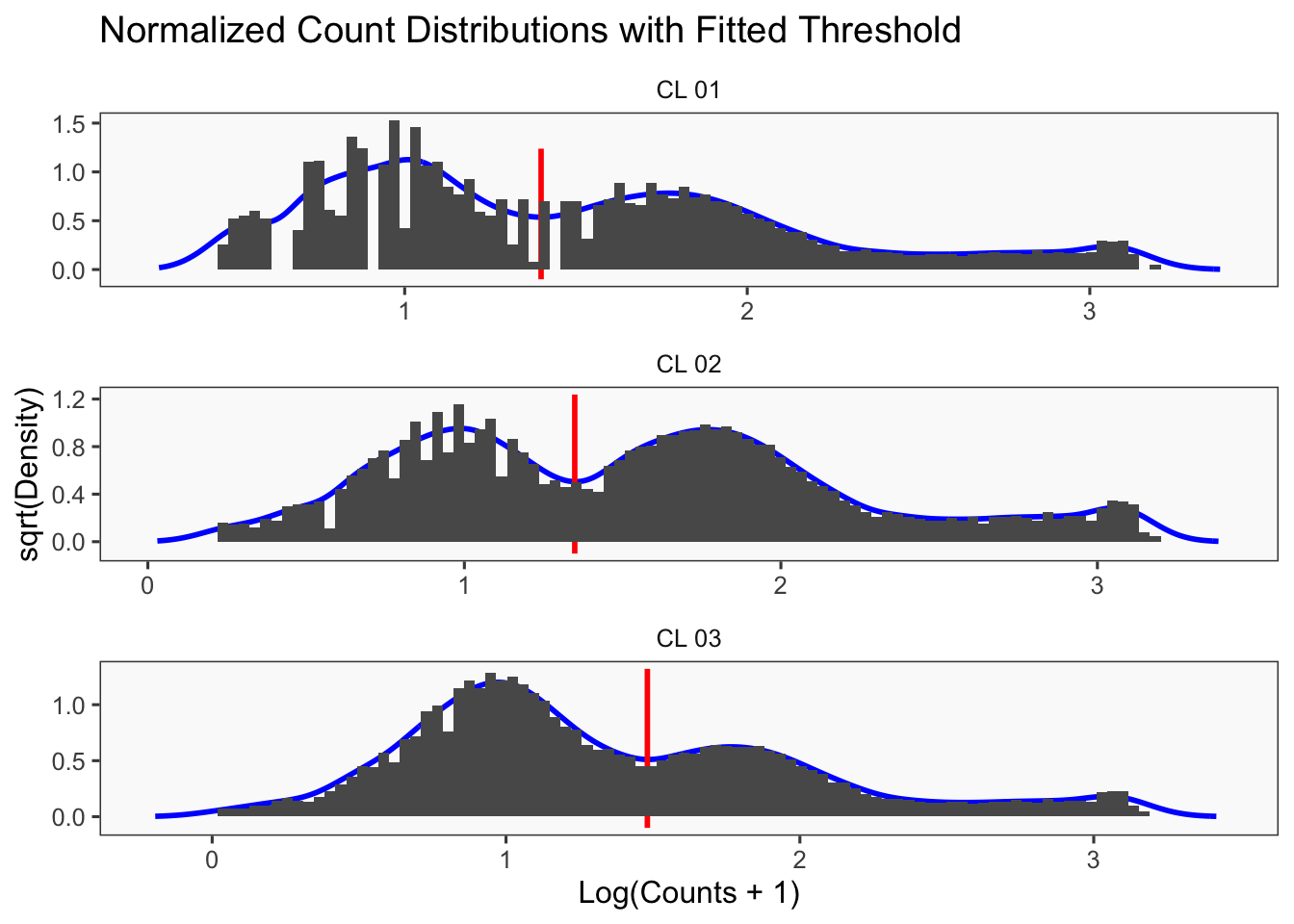
[1] "Smoothing parameter j = 5"
[1] "Smoothing parameter j = 10"
[1] "Smoothing parameter j = 15"
[1] "Smoothing parameter j = 20"
[1] "Smoothing parameter j = 25"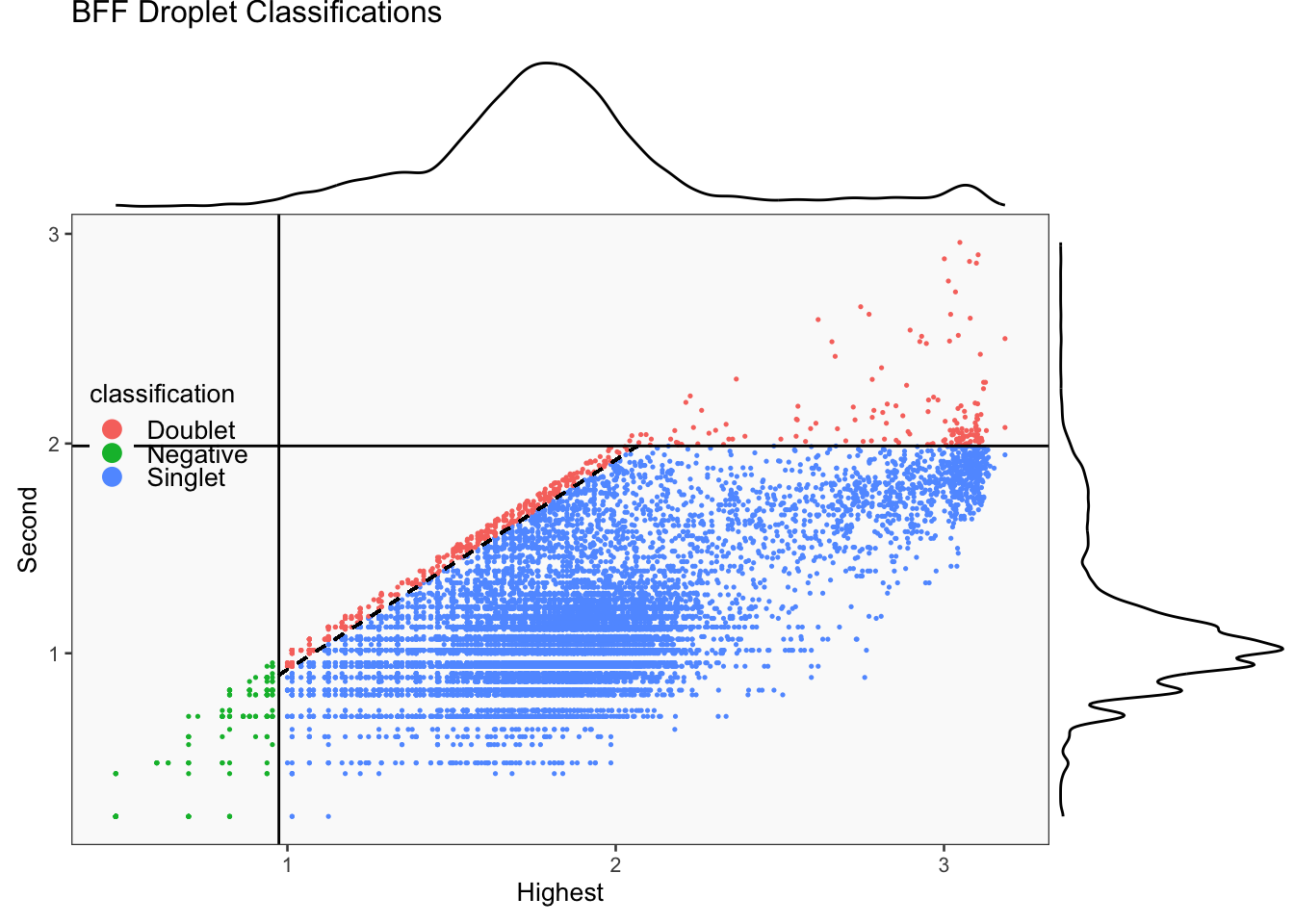
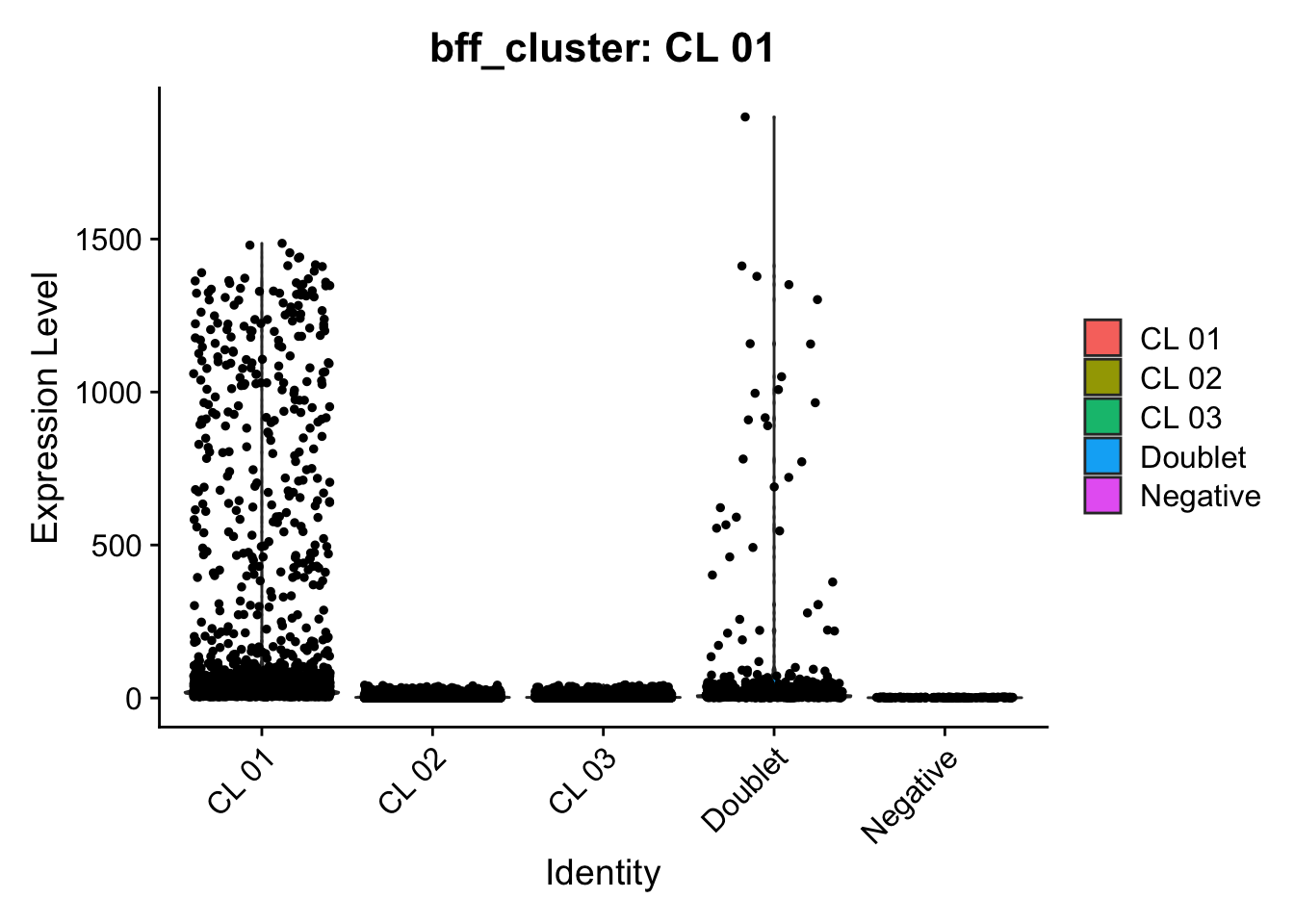
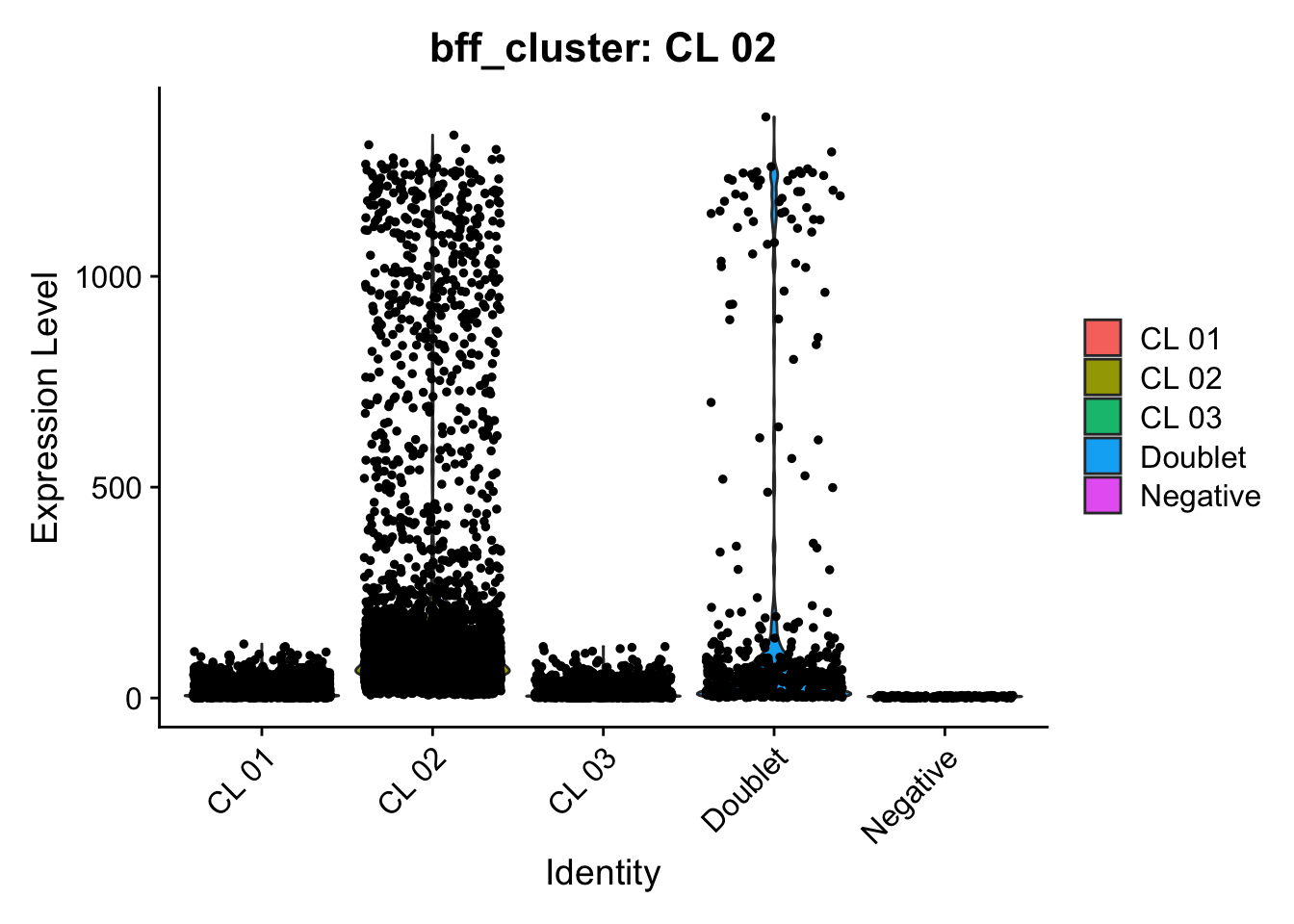
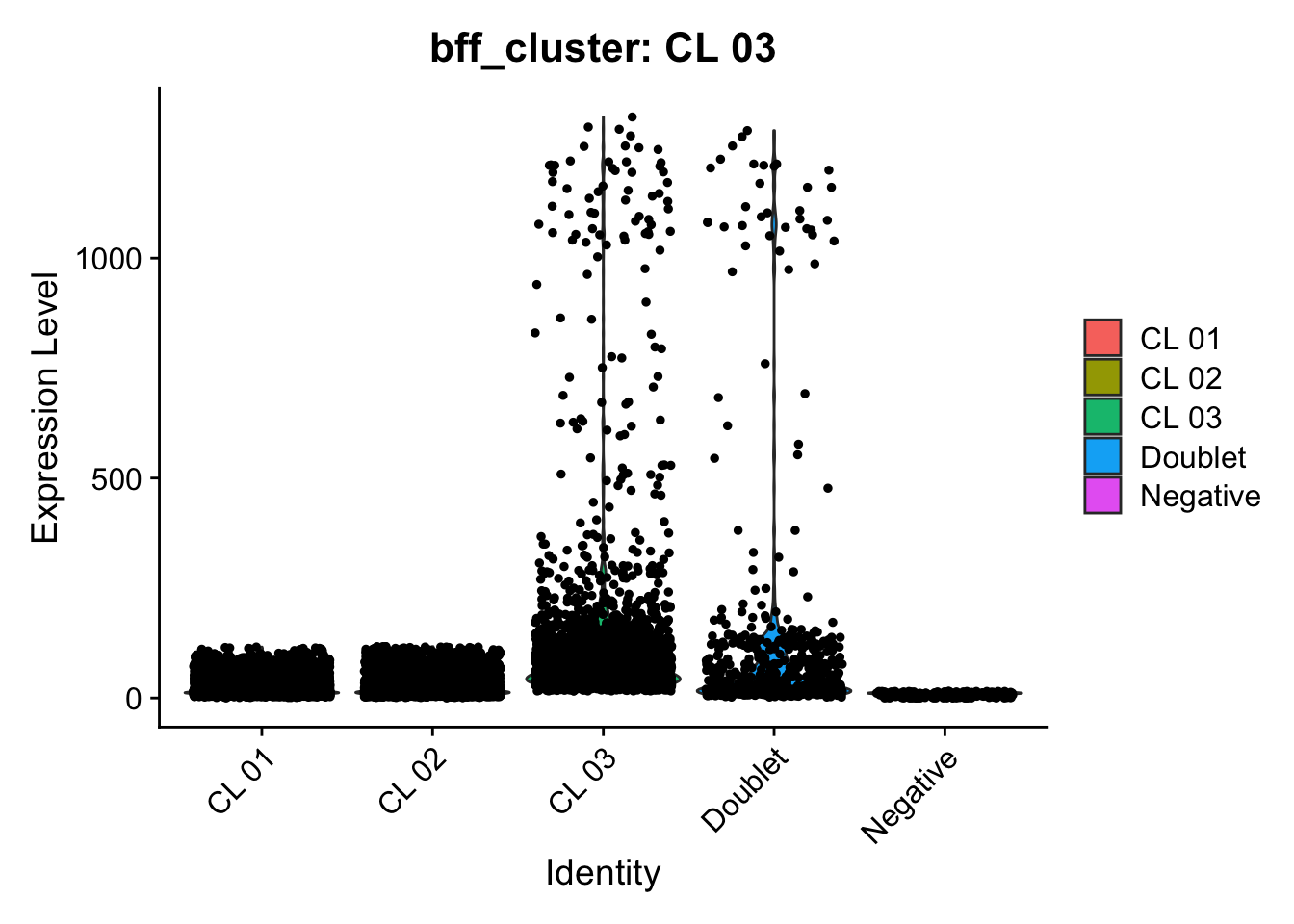
[1] "Generating consensus calls"
[1] "Consensus calls will be generated using: bff_raw,bff_cluster"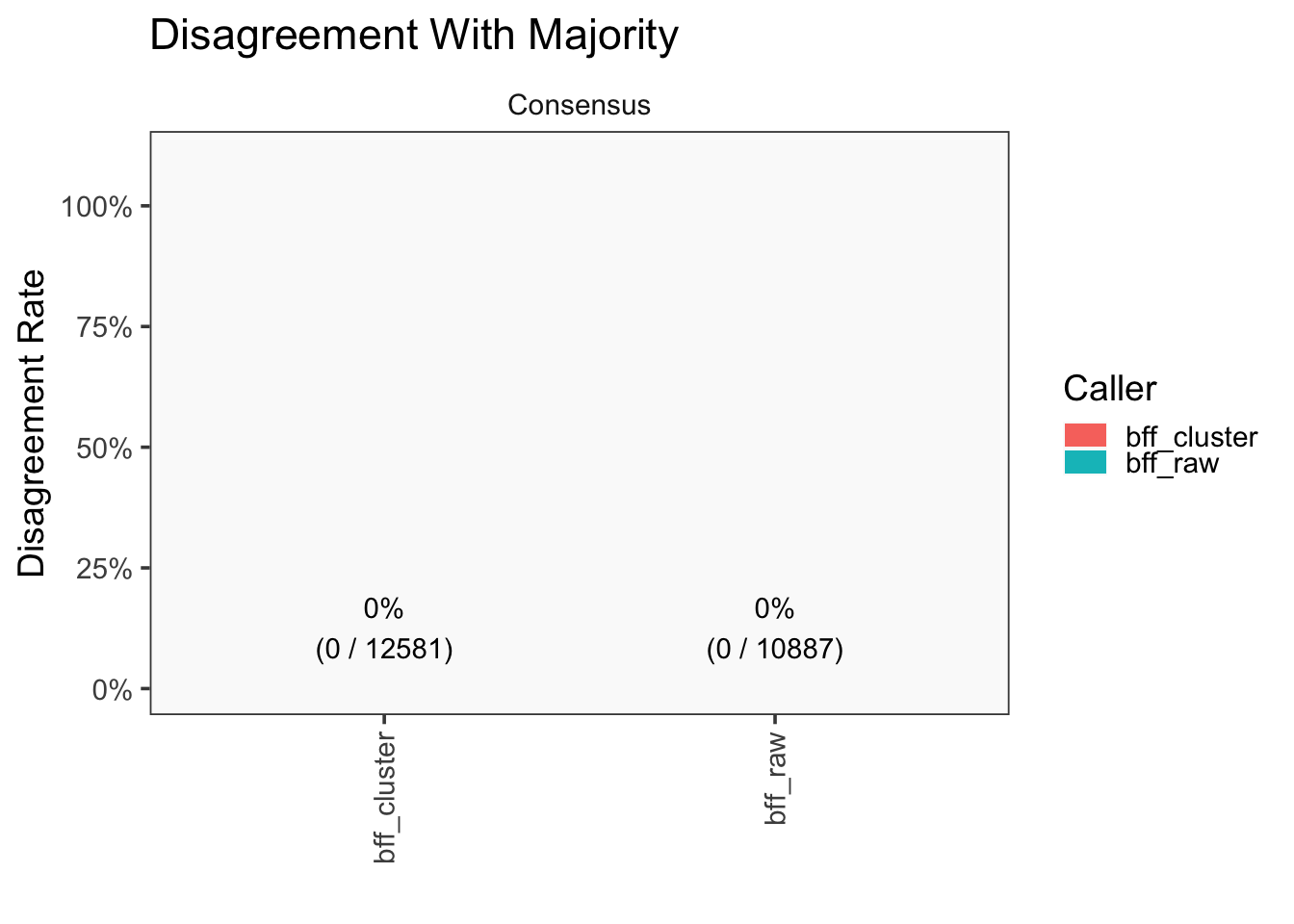
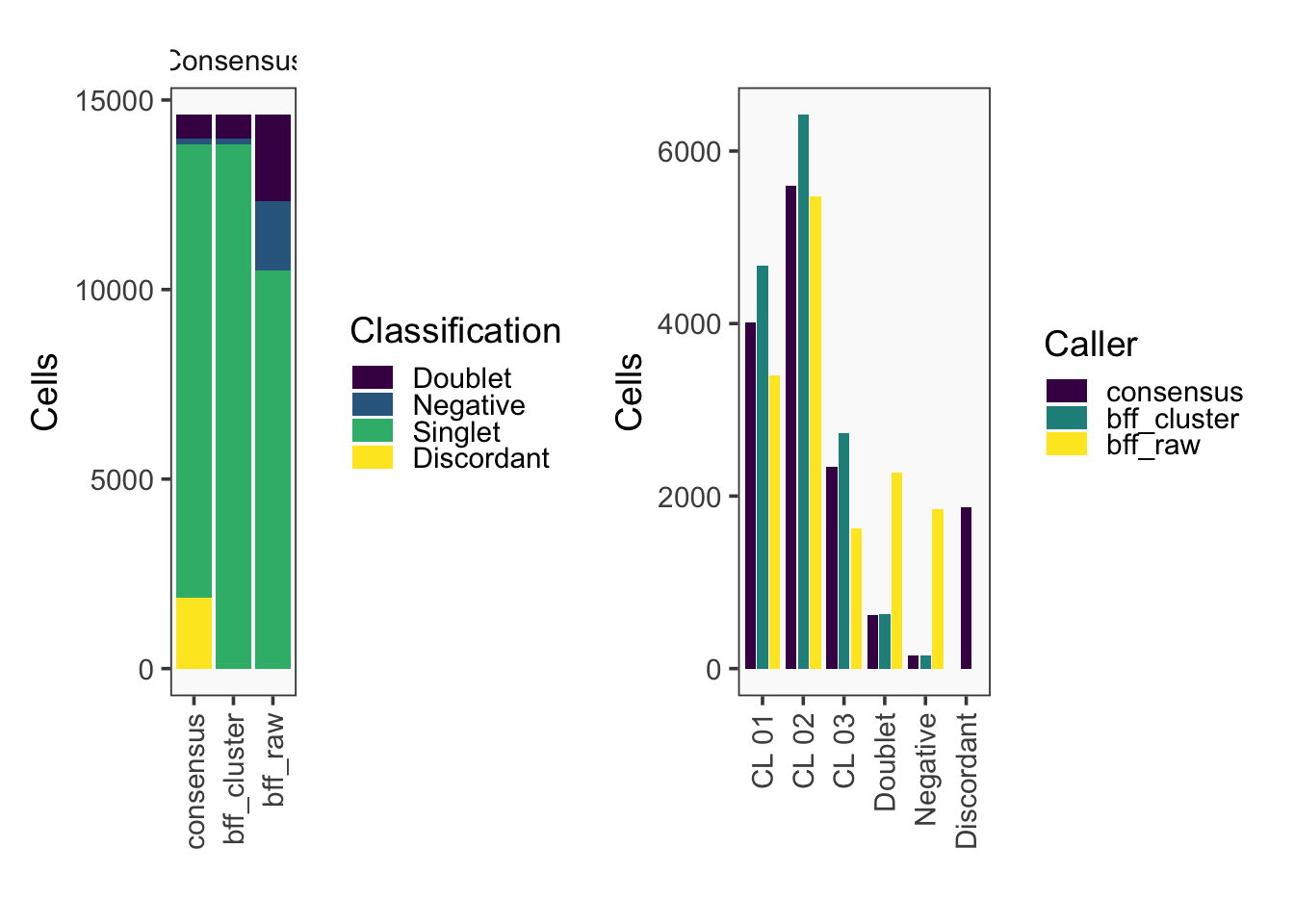
[1] "Total concordant: 12731"
[1] "Total discordant: 1876 (12.84%)"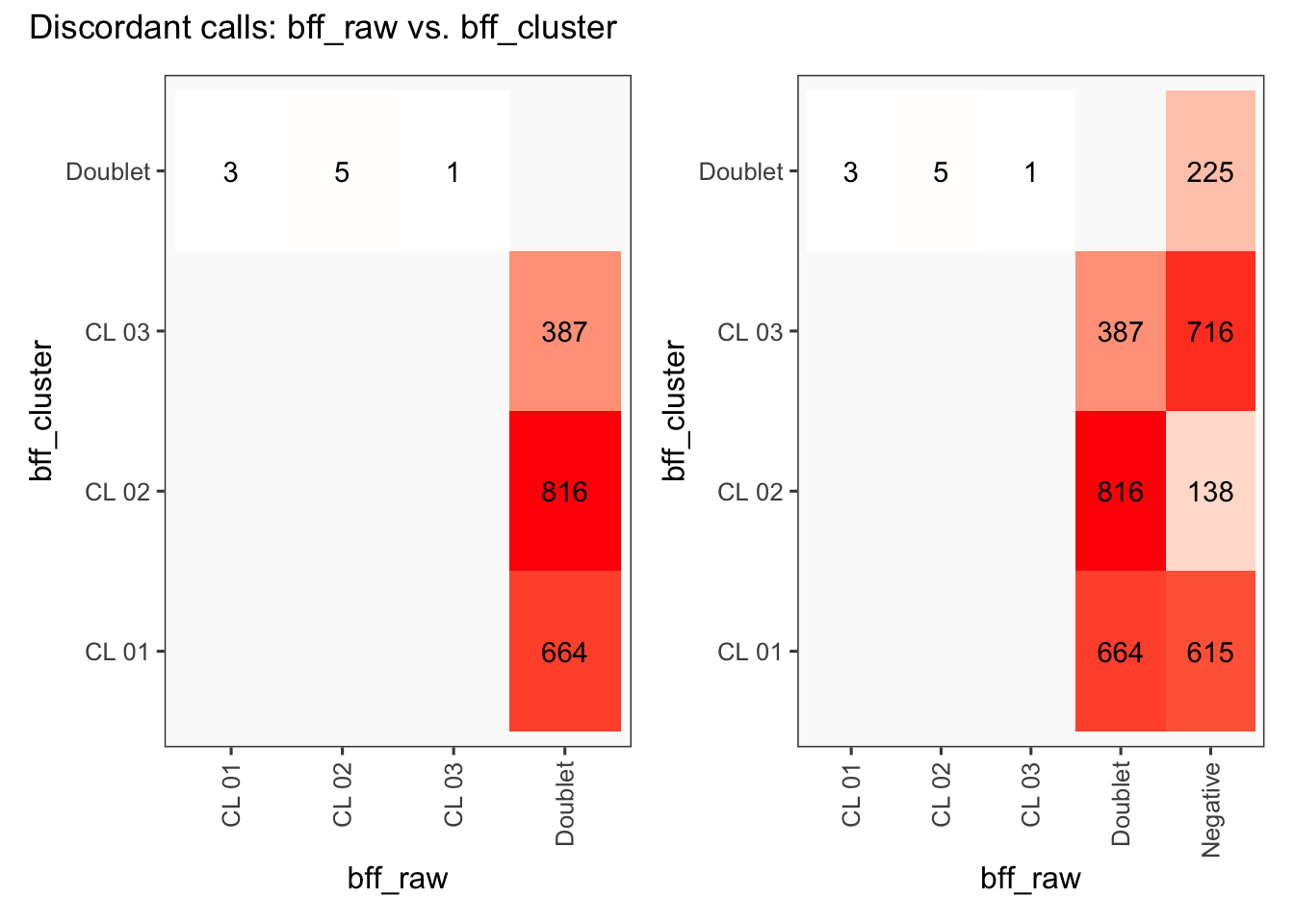
seu_lmo_c2$BFF_raw_calls <- cellhashR_calls$bff_raw
seu_lmo_c2$BFF_cluster_calls <- cellhashR_calls$bff_cluster
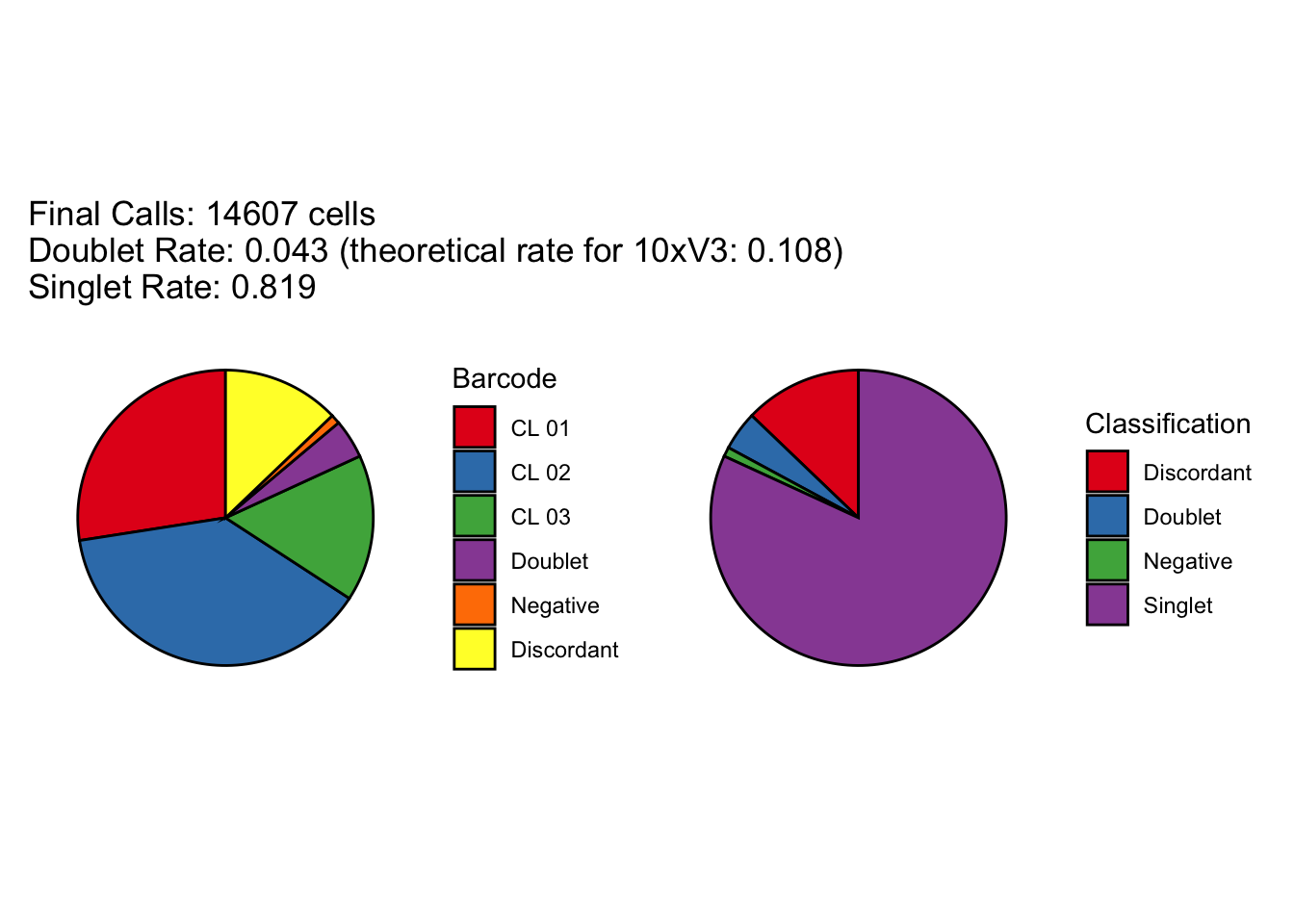
cellhashR_calls <- GenerateCellHashingCalls(barcodeMatrix = lmo_counts_c3, methods = c("bff_raw", "bff_cluster"), doTSNE = FALSE, doHeatmap = FALSE)[1] "Starting BFF"
[1] "rows dropped for low counts: 0 of 3"
[1] "Running BFF_raw"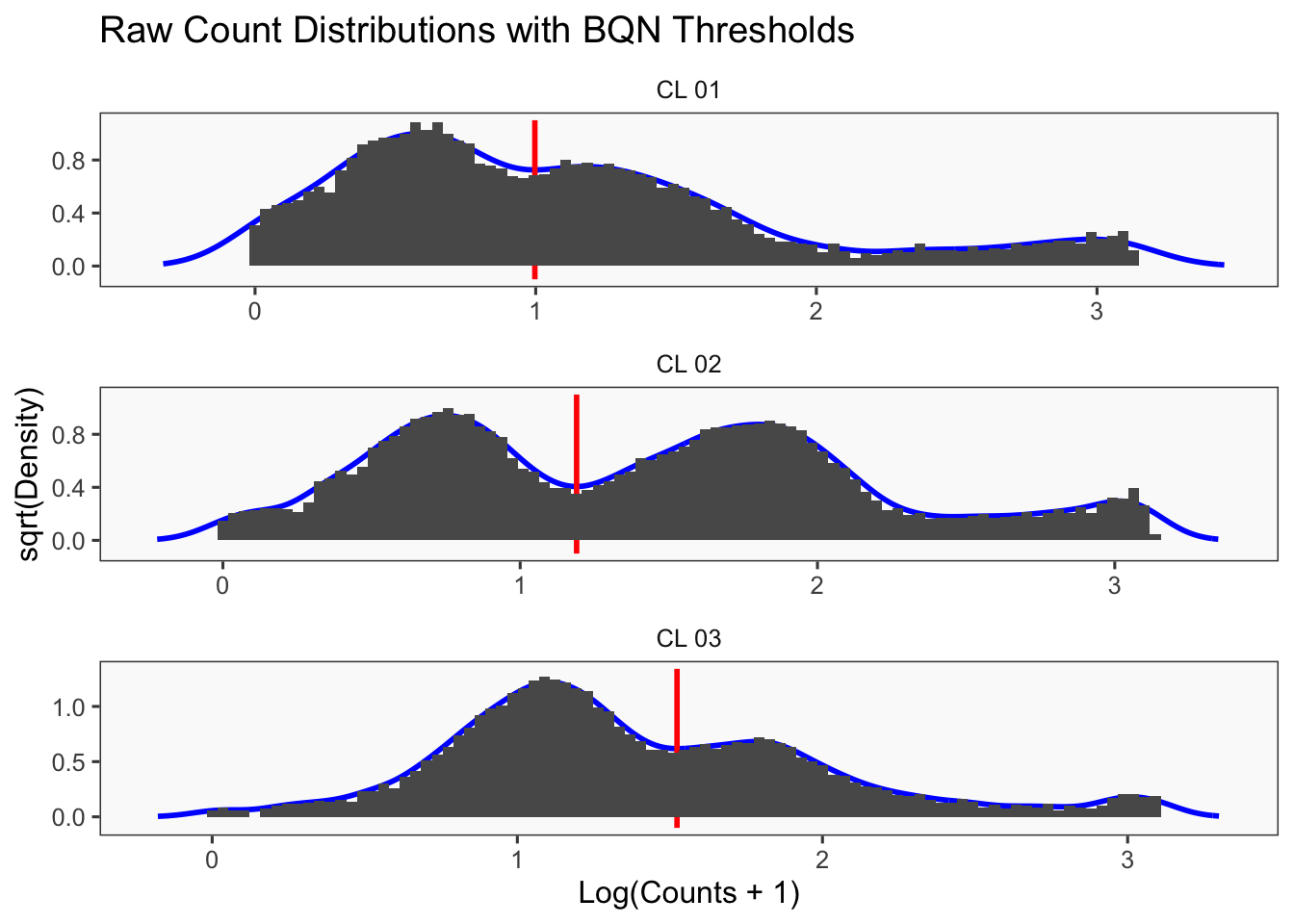
[1] "Thresholds:"
[1] "CL 03: 33.3627128550485"
[1] "CL 02: 15.4824334840404"
[1] "CL 01: 9.93179643719681"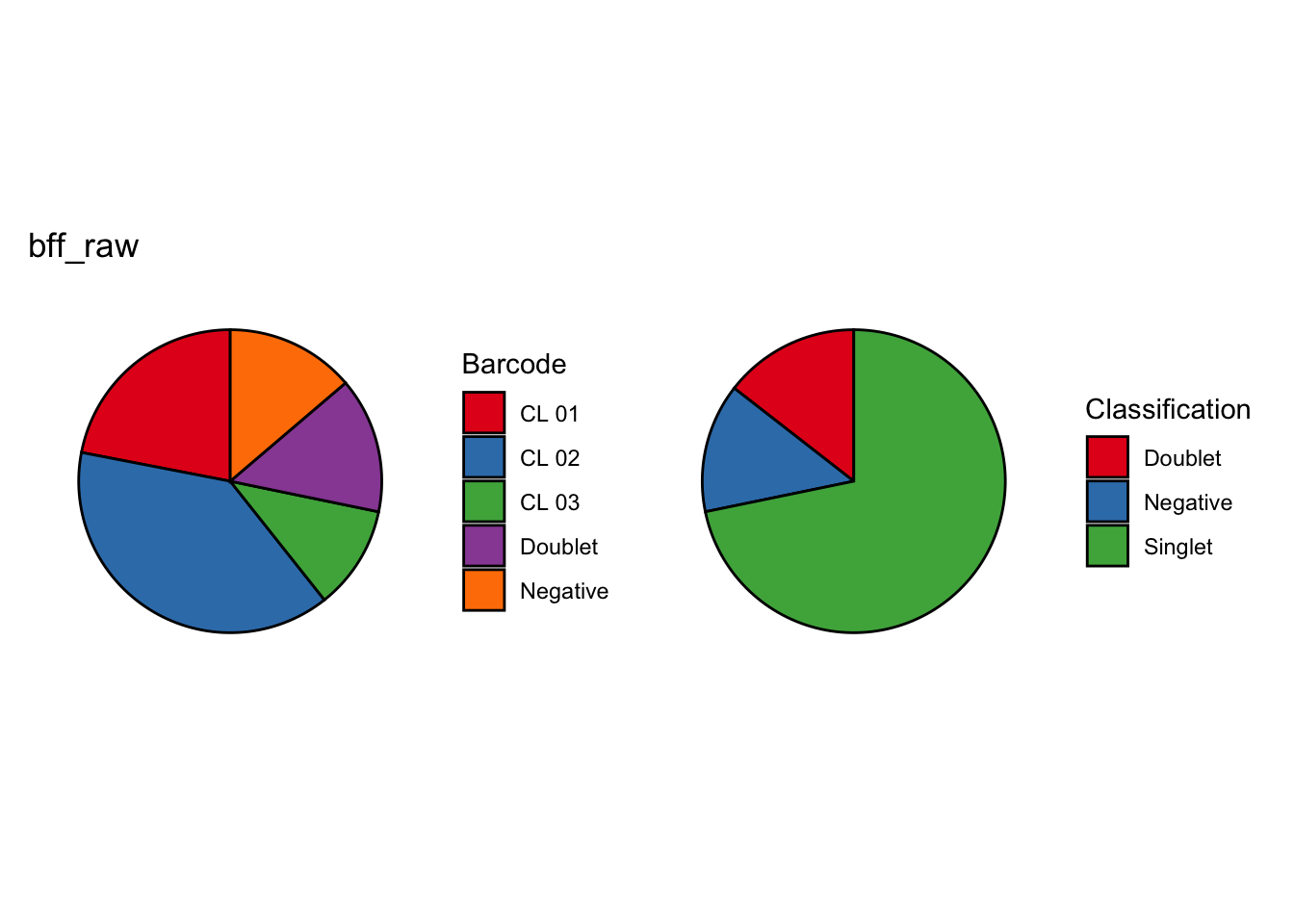
[1] "Starting BFF"
[1] "rows dropped for low counts: 0 of 3"
[1] "Running BFF_cluster"
[1] "Doublet threshold: 0.05"
[1] "Neg threshold: 0.05"
[1] "Min distance as fraction of distance between peaks: 0.1"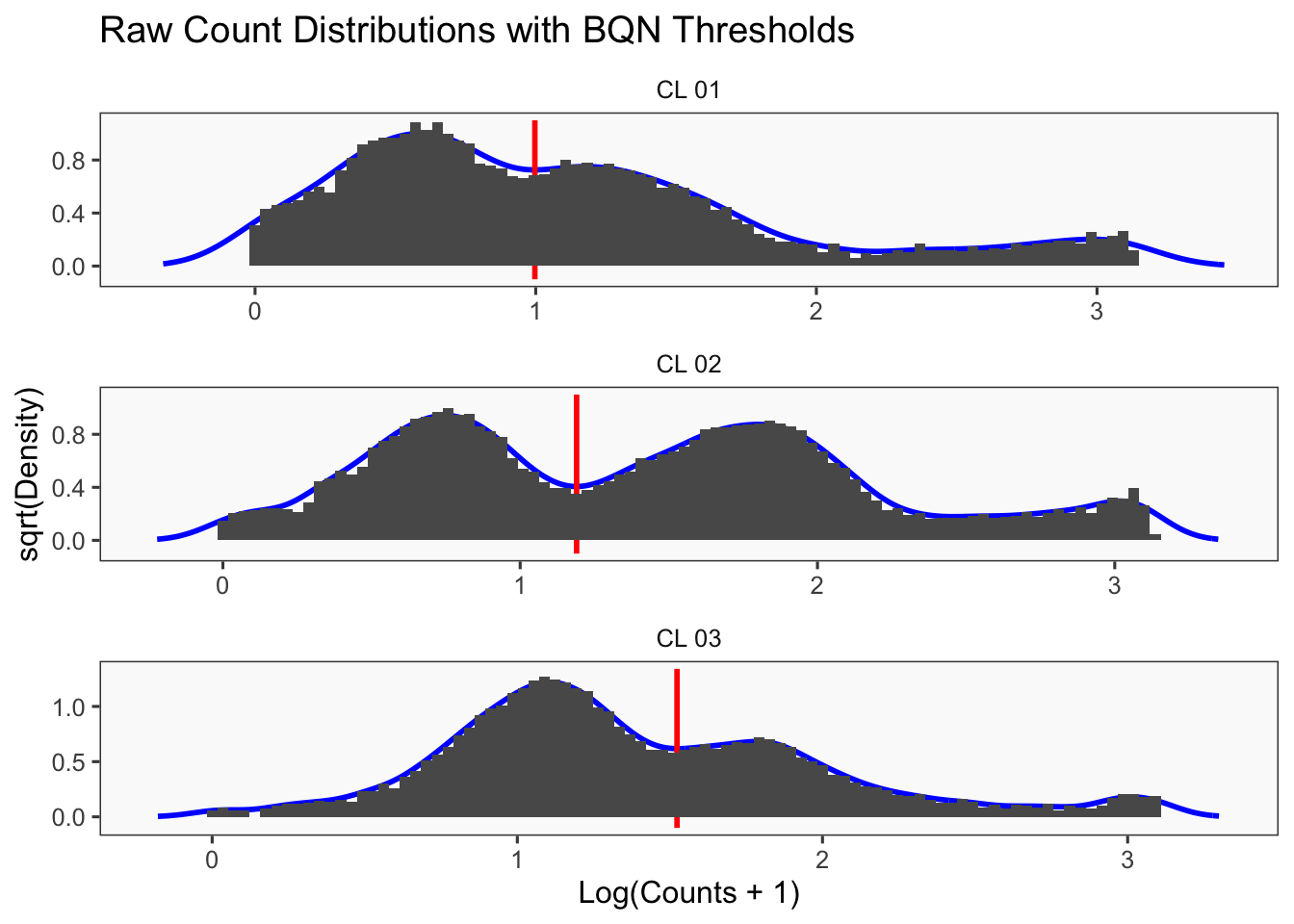
[1] "Thresholds:"
[1] "CL 03: 33.3627128550485"
[1] "CL 02: 15.4824334840404"
[1] "CL 01: 9.93179643719681"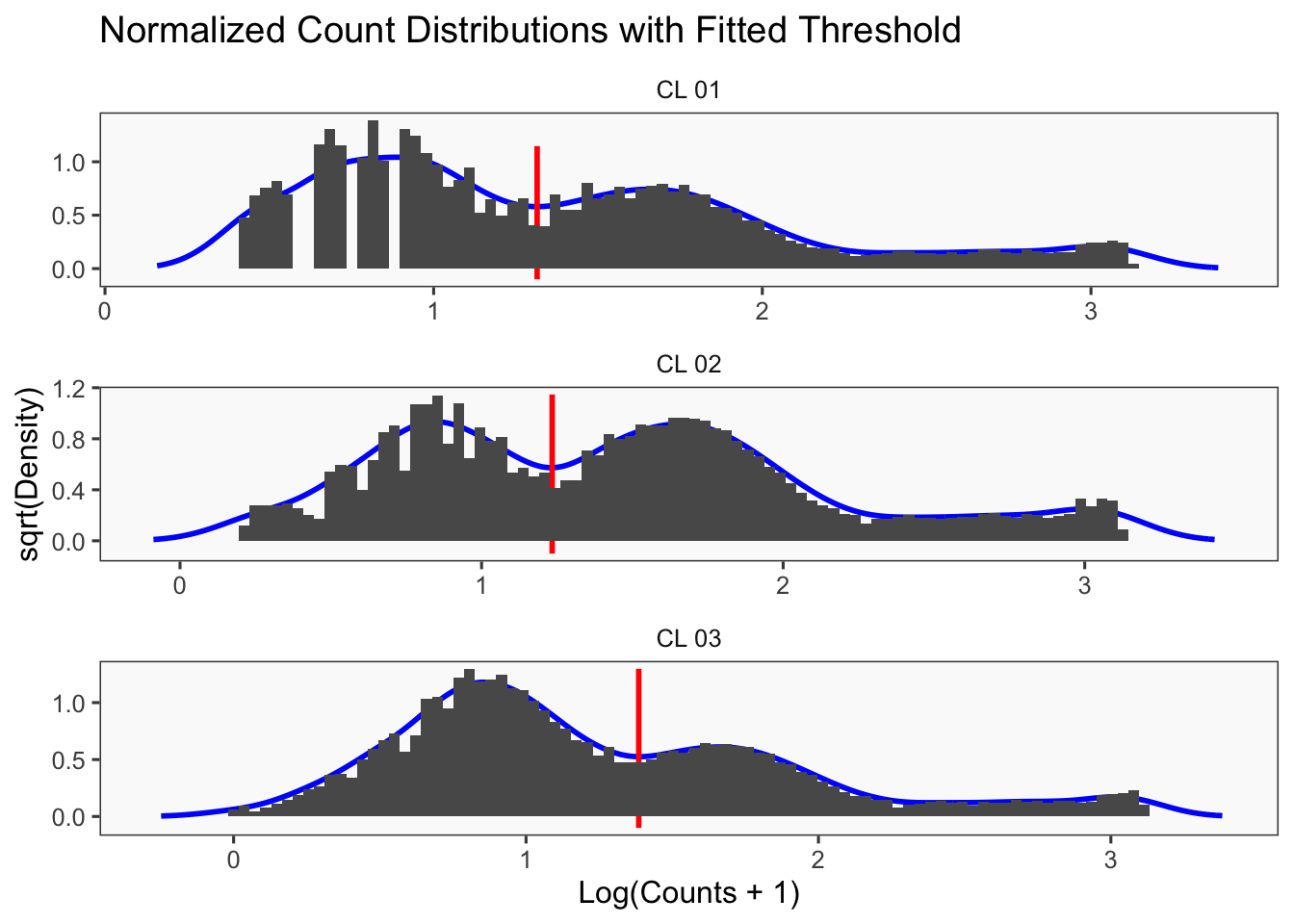
[1] "Smoothing parameter j = 5"
[1] "Smoothing parameter j = 10"
[1] "Smoothing parameter j = 15"
[1] "Smoothing parameter j = 20"
[1] "Smoothing parameter j = 25"
[1] "Smoothing parameter j = 30"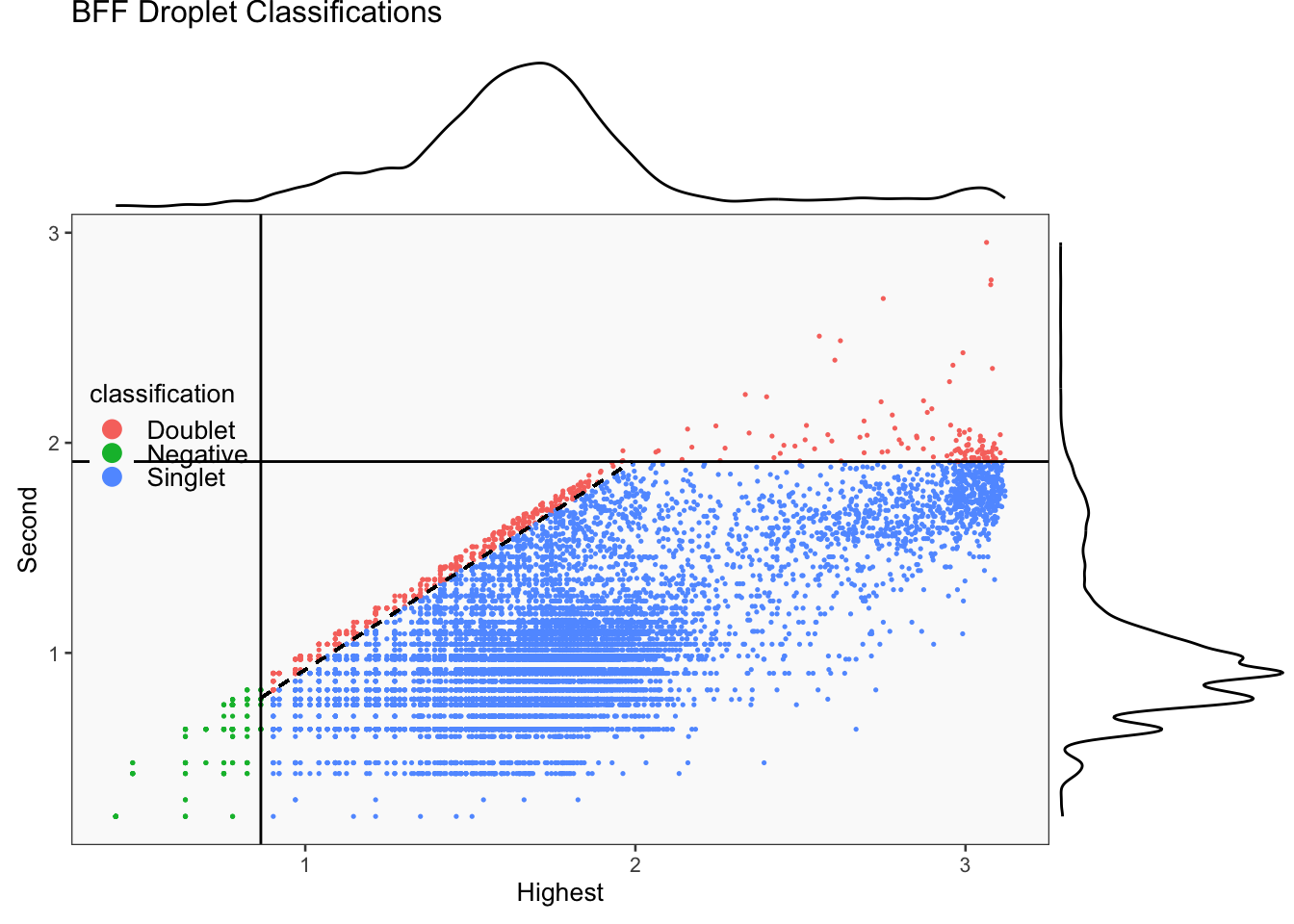
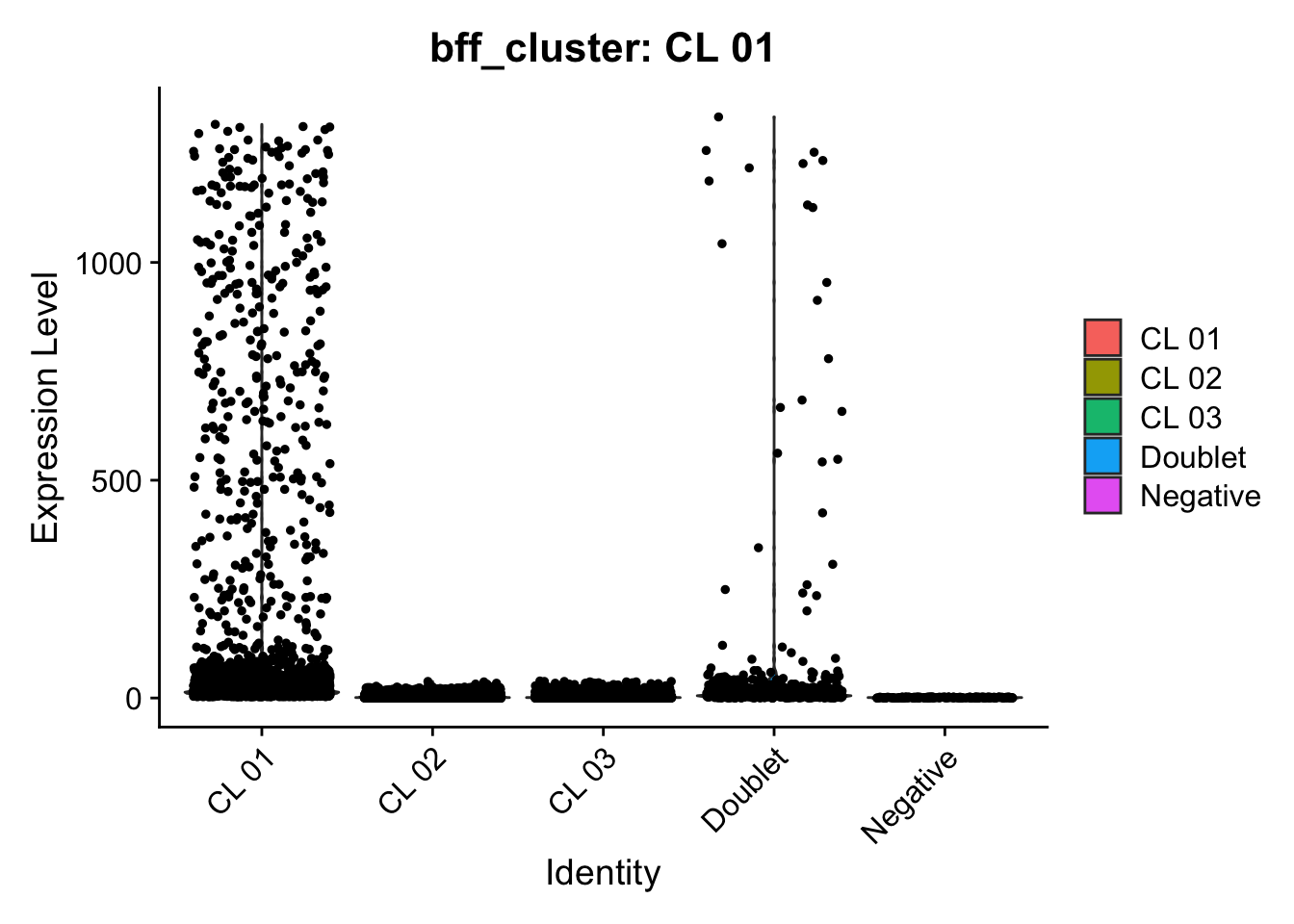
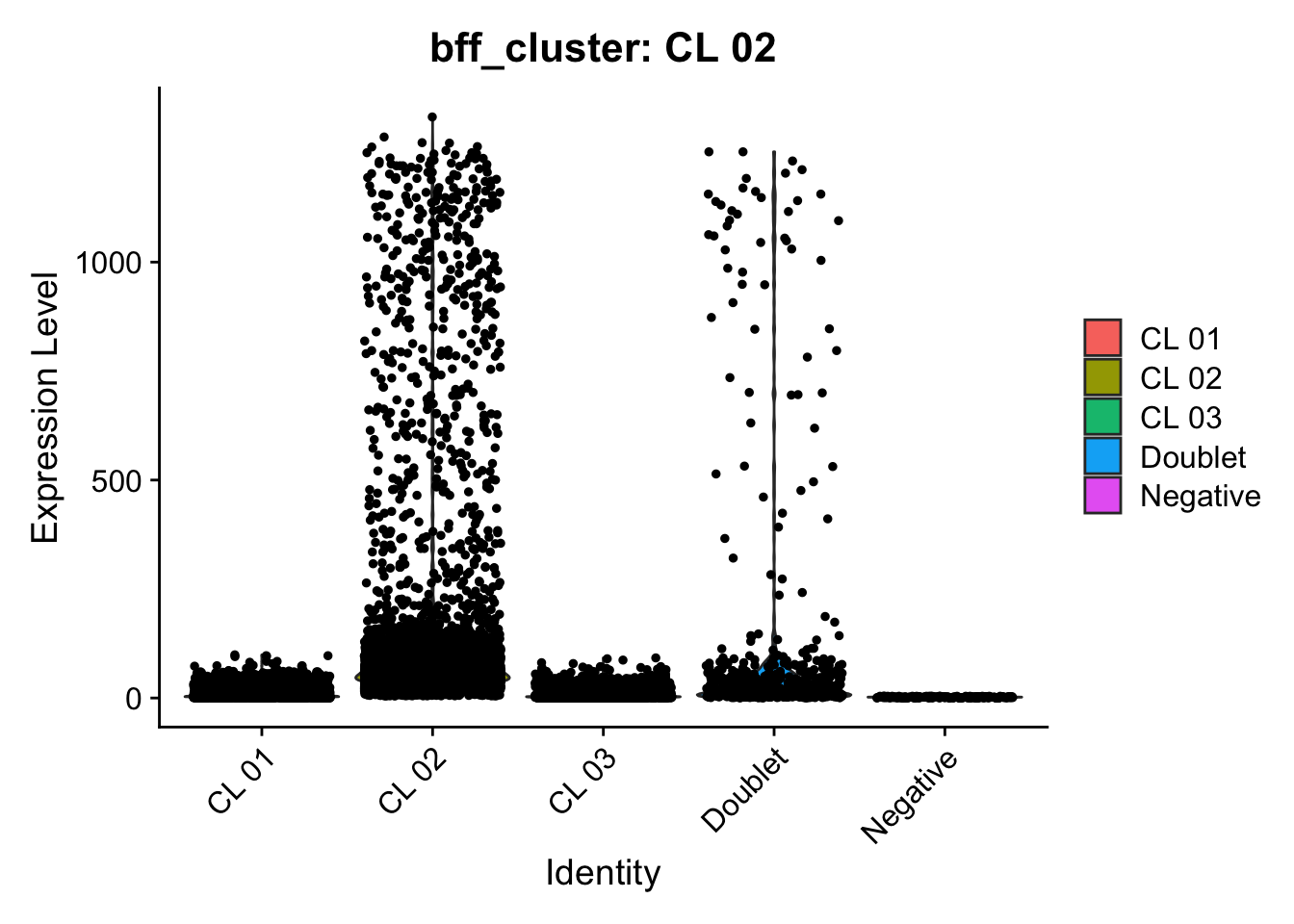
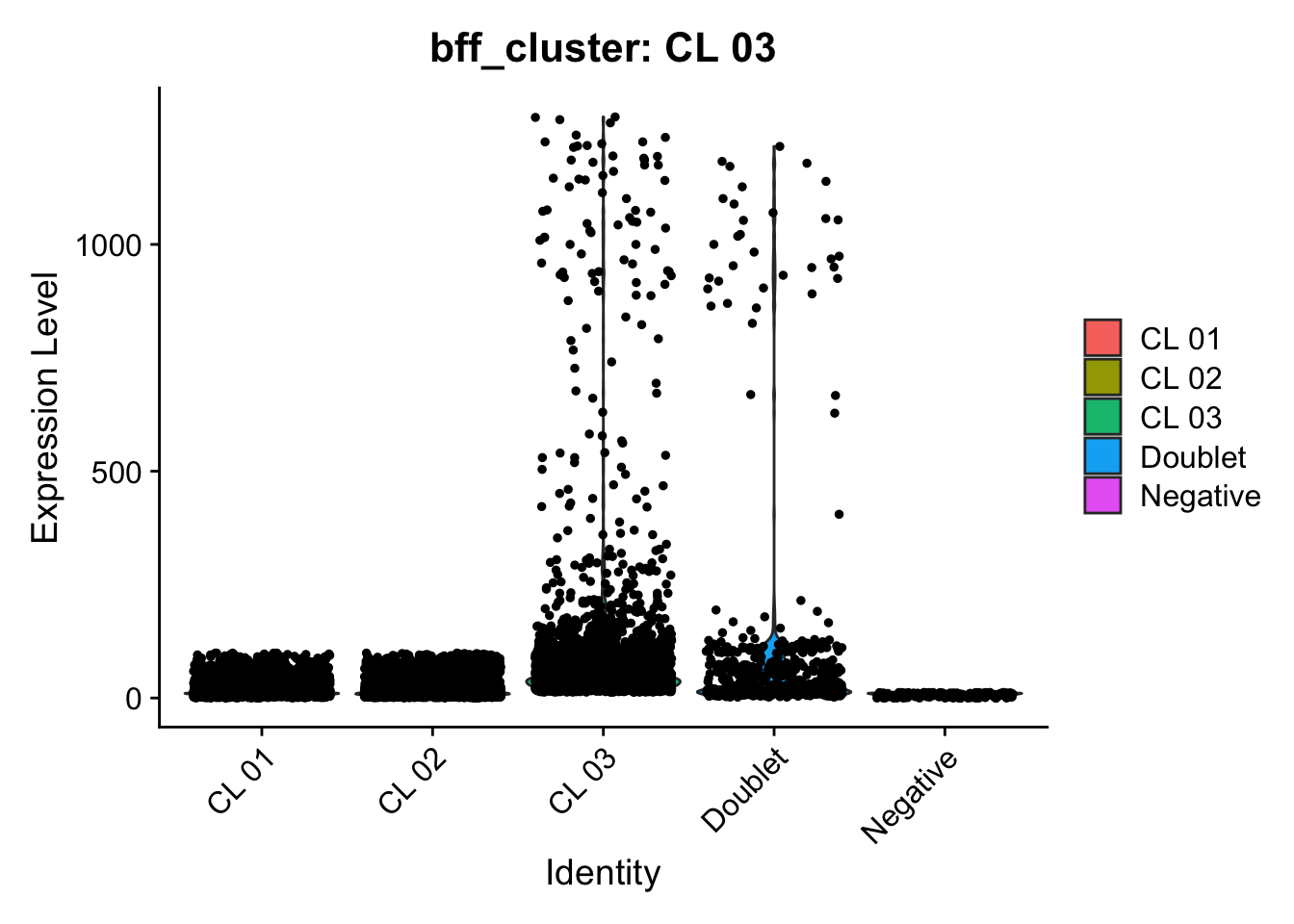
[1] "Generating consensus calls"
[1] "Consensus calls will be generated using: bff_raw,bff_cluster"
[1] "Total concordant: 12965"
[1] "Total discordant: 1807 (12.23%)"
seu_lmo_c3$BFF_raw_calls <- cellhashR_calls$bff_raw
seu_lmo_c3$BFF_cluster_calls <- cellhashR_calls$bff_cluster###demuxmix
This function turns the output of demuxmix into something consistent with the other methods
demuxmix_calls_consistent <- function(seurat_object, model = "naive", hto_list) {
hto_counts <- as.matrix(GetAssayData(seurat_object[["HTO"]], slot = "counts"))
dmm <- demuxmix(hto_counts, model = model)
dmm_calls <- dmmClassify(dmm)
calls_out <- case_when(dmm_calls$HTO %in% hto_list ~ dmm_calls$HTO,
!dmm_calls$HTO %in% hto_list ~ case_when(
dmm_calls$Type == "multiplet" ~ "Doublet",
dmm_calls$Type %in% c("negative", "uncertain") ~ "Negative")
)
return(as.factor(calls_out))
}seu_lmo_c1$demuxmix_calls <- demuxmix_calls_consistent(seu_lmo_c1, hto_list = LMO_list)
seu_lmo_c2$demuxmix_calls <- demuxmix_calls_consistent(seu_lmo_c2, hto_list = LMO_list)
seu_lmo_c3$demuxmix_calls <- demuxmix_calls_consistent(seu_lmo_c3, hto_list = LMO_list)Re-merge back into single seurat object
seu_lmo_c1$capture <- "capture 1"
seu_lmo_c2$capture <- "capture 2"
seu_lmo_c3$capture <- "capture 3"
seu_lmo <- merge(seu_lmo_c1, c(seu_lmo_c2, seu_lmo_c3))Save Seurat objects with all the hashtag assignments
saveRDS(seu_lmo, here("data", "cell_line_data", "lmo_all_methods.SEU.rds"))Making plots
Compute F-scores
We compute the F-score of each of the possible singlet assignments.
#Helper function
calculate_HTO_fscore <- function(seurat_object, donor_hto_list, method) {
calls <- seurat_object[[method]]
f <- NULL
for (HTO in donor_hto_list) {
tp <- sum(calls == HTO & donor_hto_list[seurat_object$genetic_donor] == HTO) #True positive rate
fp <- sum(calls == HTO & donor_hto_list[seurat_object$genetic_donor] != HTO) #False positive rate
fn <- sum(calls != HTO & donor_hto_list[seurat_object$genetic_donor] == HTO) #False negative rate
f <- c(f, tp / (tp + 0.5 * (fp + fn)))
}
# f <- c(f, median(f)) #Add median F score
f <- c(f, mean(f)) #Add mean F score
names(f) <- c(donor_hto_list, "Average")
return(f)
}Compare F scores for the methods.
Fscore_hashedDrops <- calculate_HTO_fscore(seu_lmo, donor_LMO_list[1:3], "hashedDrops_calls")
Fscore_hashedDrops_default <- calculate_HTO_fscore(seu_lmo, donor_LMO_list[1:3], "hashedDrops_default_calls")
Fscore_HTODemux <- calculate_HTO_fscore(seu_lmo, donor_LMO_list[1:3], "HTODemux_calls")
Fscore_GMMDemux <- calculate_HTO_fscore(seu_lmo, donor_LMO_list[1:3], "GMMDemux_calls")
Fscore_deMULTIplex <- calculate_HTO_fscore(seu_lmo, donor_LMO_list[1:3], "deMULTIplex_calls")
Fscore_BFF_raw <- calculate_HTO_fscore(seu_lmo, donor_LMO_list[1:3], "BFF_raw_calls")
Fscore_BFF_cluster <- calculate_HTO_fscore(seu_lmo, donor_LMO_list[1:3], "BFF_cluster_calls")
Fscore_demuxmix <- calculate_HTO_fscore(seu_lmo, donor_LMO_list[1:3], "demuxmix_calls")
Fscore_hashsolo <- calculate_HTO_fscore(seu_lmo, donor_LMO_list[1:3], "hashsolo_calls")Fscore_matrix <- data.frame("LMO" = c(LMO_list[1:3], "Mean"),
"hashedDrops" = Fscore_hashedDrops,
"hashedDrops_default" = Fscore_hashedDrops_default,
"HashSolo" = Fscore_hashsolo,
"HTODemux" = Fscore_HTODemux,
"GMM_Demux" = Fscore_GMMDemux,
"deMULTIplex" = Fscore_deMULTIplex,
"BFF_raw" = Fscore_BFF_raw,
"BFF_cluster" = Fscore_BFF_cluster,
"demuxmix" = Fscore_demuxmix)
#Removing average information for this data set
Fscore_matrix = Fscore_matrix[1:3,]Fscore_matrix %>%
pivot_longer(cols = c("hashedDrops", "hashedDrops_default", "HashSolo", "HTODemux", "GMM_Demux", "deMULTIplex", "BFF_raw", "BFF_cluster", "demuxmix"),
names_to = "method",
values_to = "Fscore") -> Fscore_matrixp1 <- heatmap(Fscore_matrix,
.row = method,
.column = LMO,
.value = Fscore,
column_title = "F-score - Cell line data",
cluster_rows = TRUE,
row_names_gp = gpar(fontsize = 10),
show_row_dend = FALSE,
row_names_side = "left",
row_title = "",
cluster_columns = FALSE,
column_names_gp = gpar(fontsize = 10),
palette_value = plasma(3)) %>%
wrap_heatmap()tidyHeatmap says: (once per session) from release 1.7.0 the scaling is set to "none" by default. Please use scale = "row", "column" or "both" to apply scalingp1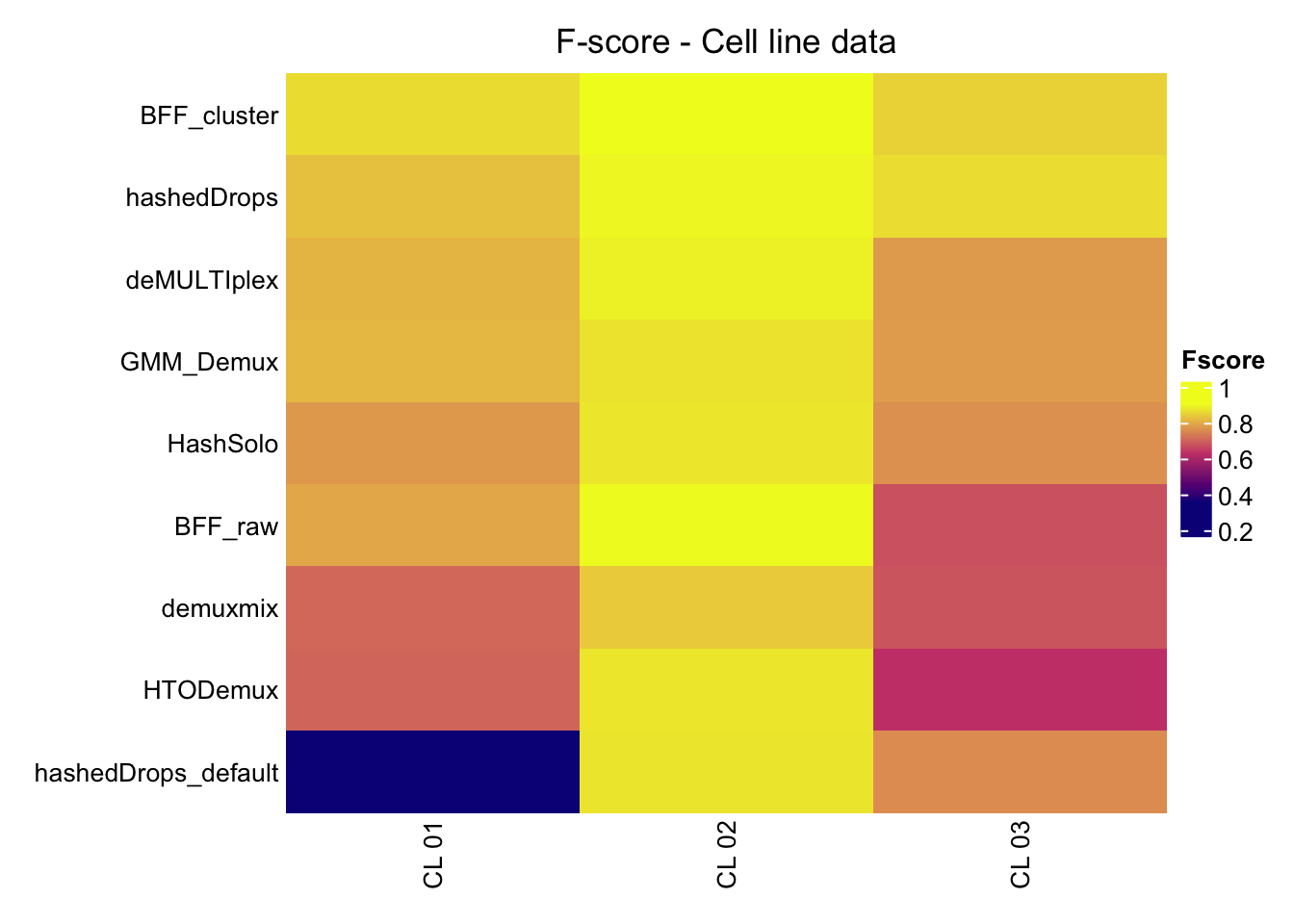
#ggsave(here("paper_latex", "figures", "CL_Fscore.png"),
# p1,
# device = "png",
# width = 6, height = 4,
# units = "in",
# dpi = 350
# )Doublet-to-singlet assignments
method_calls <- c("hashedDrops_calls",
"hashedDrops_default_calls",
"hashsolo_calls",
"HTODemux_calls",
"GMMDemux_calls",
"deMULTIplex_calls",
"BFF_raw_calls",
"BFF_cluster_calls",
"demuxmix_calls")
doublets <- seu_lmo[, seu_lmo$genetic_donor == "Doublet"]
doublet_doublet <- NULL
doublet_negative <- NULL
doublet_singlet <- NULL
for (method in method_calls) {
doublet_doublet <- c(doublet_doublet, sum(seu_lmo$genetic_donor == "Doublet" & seu_lmo[[method]] == "Doublet") / sum(seu_lmo$genetic_donor == "Doublet"))
doublet_negative <- c(doublet_negative, sum(seu_lmo$genetic_donor == "Doublet" & seu_lmo[[method]] == "Negative") / sum(seu_lmo$genetic_donor == "Doublet"))
doublet_singlet <- c(doublet_singlet, sum(seu_lmo$genetic_donor == "Doublet" & Reduce("|", lapply(LMO_list[1:3], function(x) seu_lmo[[method]] == x))) / sum(seu_lmo$genetic_donor == "Doublet"))
}
names(doublet_doublet) <- method_calls
names(doublet_negative) <- method_calls
names(doublet_singlet) <- method_callsdoublet_assignments <- data.frame("method" = c("hashedDrops",
"hashedDrops (default)",
"HashSolo", "HTODemux",
"GMM-Demux",
"deMULTIplex",
"BFF_raw",
"BFF_cluster",
"demuxmix"),
"Doublets" = doublet_doublet,
"Negative" = doublet_negative,
"Singlet" = doublet_singlet) %>%
pivot_longer(cols = c("Doublets", "Negative", "Singlet"),
names_to = "assignment",
values_to = "fraction")doublet_colours <- c("black", "gray60", "firebrick1")
p2 <- ggplot(doublet_assignments %>%
mutate(method = factor(method, levels = c("hashedDrops (default)",
"HTODemux",
"demuxmix",
"BFF_raw",
"HashSolo",
"GMM-Demux",
"deMULTIplex",
"hashedDrops",
"BFF_cluster")))) +
geom_bar(aes(x = method, y = fraction, fill = assignment),
stat = "identity") +
ggtitle("Cell line data (4945 doublets)") +
ylim(0, 1) +
scale_fill_manual(values = doublet_colours) +
theme(axis.ticks.x = element_blank(),
axis.title.x = element_blank(),
axis.title.y = element_blank(),
plot.title = element_text(size = 10)
) + coord_flip()
p2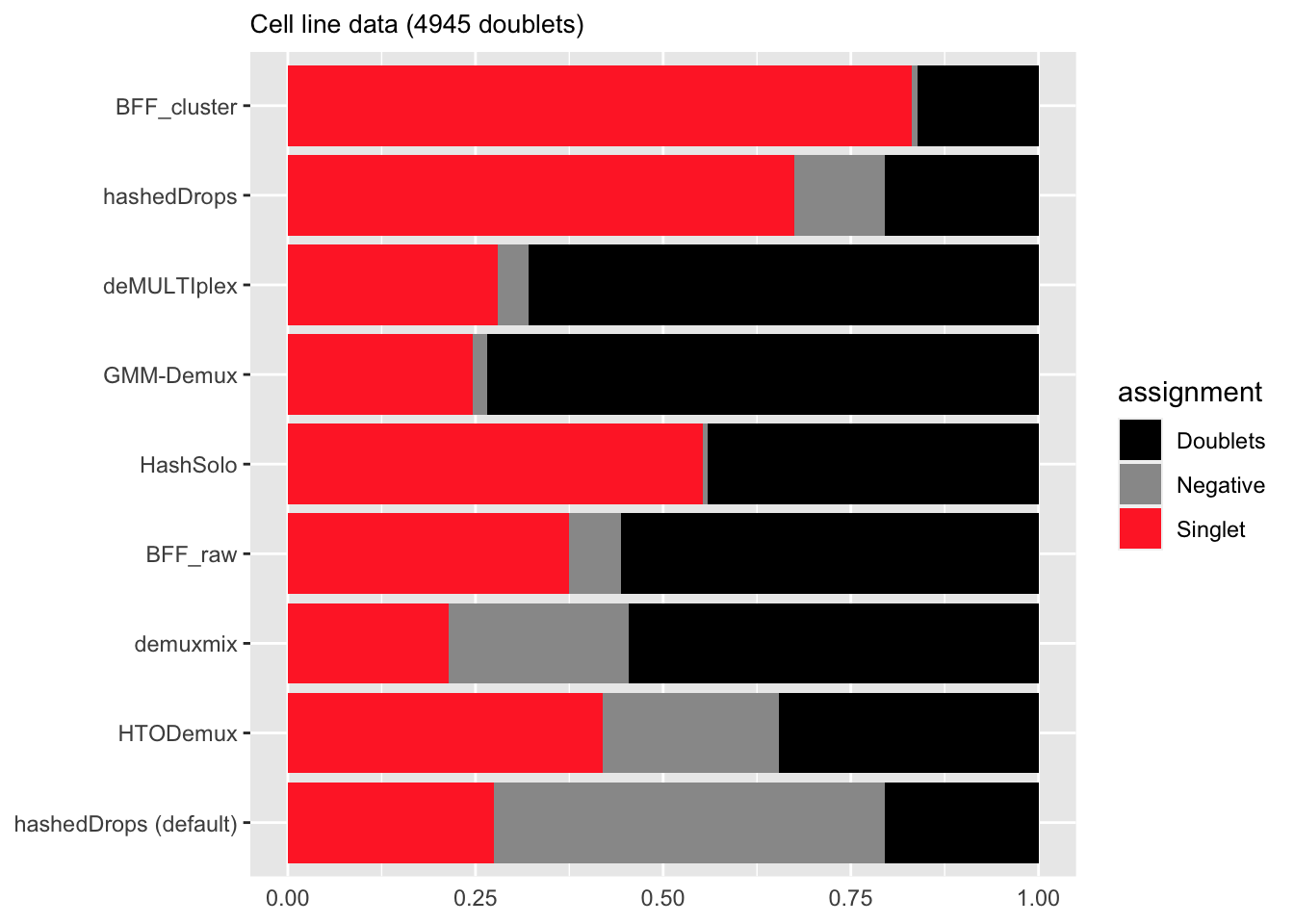
#ggsave(here("paper_latex", "figures", "CL_doublet_assignments.png"),
# p2,
# device = "png",
# width = 6, height = 4,
# units = "in",
# dpi = 350
# )How many droplets in the third peak?
#df <- as.data.frame(t(seu_lmo[["HTO"]]@counts))
#colnames(df) <- gsub("_", " ", LMO_donor_list[colnames(df)])
#df %>%
# pivot_longer(cols = starts_with("donor")) %>%
# mutate(logged = log10(value + 1)) %>%
# ggplot(aes(x = logged)) +
# xlab("log10(counts)") +
# xlim(0.1,4) +
# geom_density(adjust = 2) +
# facet_wrap(~name, scales = "fixed", ncol = 3) -> p1
#p1#table(seu_lmo$genetic_donor)
#sum(log10(seu_lmo[["HTO"]]@counts[1,]) > 2)
#sum(log10(seu_lmo[["HTO"]]@counts[2,]) > 2.5)
#sum(log10(seu_lmo[["HTO"]]@counts[3,]) > 2.5)
sessionInfo()R version 4.2.2 (2022-10-31)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS Ventura 13.0.1
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.2-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.2-arm64/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] grid stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] Matrix_1.5-4 viridis_0.6.3
[3] viridisLite_0.4.2 tidyHeatmap_1.8.1
[5] ComplexHeatmap_2.14.0 demuxmix_1.0.0
[7] RColorBrewer_1.1-3 cellhashR_1.0.3
[9] dittoSeq_1.10.0 speckle_0.99.7
[11] pheatmap_1.0.12 SeuratObject_4.1.3
[13] Seurat_4.3.0 scater_1.26.1
[15] scuttle_1.8.4 lubridate_1.9.2
[17] forcats_1.0.0 stringr_1.5.0
[19] purrr_1.0.1 readr_2.1.4
[21] tidyr_1.3.0 tibble_3.2.1
[23] tidyverse_2.0.0 DropletUtils_1.18.1
[25] SingleCellExperiment_1.20.1 SummarizedExperiment_1.28.0
[27] Biobase_2.58.0 GenomicRanges_1.50.2
[29] GenomeInfoDb_1.34.9 IRanges_2.32.0
[31] S4Vectors_0.36.2 BiocGenerics_0.44.0
[33] MatrixGenerics_1.10.0 matrixStats_0.63.0
[35] patchwork_1.1.2 cowplot_1.1.1
[37] ggplot2_3.4.2 janitor_2.2.0
[39] dplyr_1.1.2 BiocStyle_2.26.0
[41] here_1.0.1 workflowr_1.7.0
loaded via a namespace (and not attached):
[1] scattermore_1.0 R.methodsS3_1.8.2
[3] knitr_1.42 irlba_2.3.5.1
[5] DelayedArray_0.24.0 R.utils_2.12.2
[7] data.table_1.14.8 RCurl_1.98-1.12
[9] doParallel_1.0.17 generics_0.1.3
[11] preprocessCore_1.61.0 ScaledMatrix_1.6.0
[13] callr_3.7.3 RANN_2.6.1
[15] future_1.32.0 tzdb_0.3.0
[17] spatstat.data_3.0-1 httpuv_1.6.11
[19] xfun_0.39 hms_1.1.3
[21] jquerylib_0.1.4 evaluate_0.21
[23] promises_1.2.0.1 fansi_1.0.4
[25] dendextend_1.17.1 igraph_1.4.2
[27] DBI_1.1.3 htmlwidgets_1.6.2
[29] spatstat.geom_3.2-1 ellipsis_0.3.2
[31] bookdown_0.34 deldir_1.0-6
[33] sparseMatrixStats_1.10.0 vctrs_0.6.2
[35] Cairo_1.6-0 ROCR_1.0-11
[37] abind_1.4-5 cachem_1.0.8
[39] withr_2.5.0 ggforce_0.4.1
[41] progressr_0.13.0 sctransform_0.3.5
[43] goftest_1.2-3 cluster_2.1.4
[45] lazyeval_0.2.2 crayon_1.5.2
[47] spatstat.explore_3.2-1 edgeR_3.40.2
[49] pkgconfig_2.0.3 labeling_0.4.2
[51] tweenr_2.0.2 nlme_3.1-162
[53] vipor_0.4.5 rlang_1.1.1
[55] globals_0.16.2 lifecycle_1.0.3
[57] miniUI_0.1.1.1 rsvd_1.0.5
[59] ggrastr_1.0.1 rprojroot_2.0.3
[61] polyclip_1.10-4 lmtest_0.9-40
[63] Rhdf5lib_1.20.0 zoo_1.8-12
[65] beeswarm_0.4.0 whisker_0.4.1
[67] ggridges_0.5.4 GlobalOptions_0.1.2
[69] processx_3.8.1 png_0.1-8
[71] rjson_0.2.21 bitops_1.0-7
[73] getPass_0.2-2 R.oo_1.25.0
[75] KernSmooth_2.23-21 rhdf5filters_1.10.1
[77] ggExtra_0.10.0 DelayedMatrixStats_1.20.0
[79] shape_1.4.6 parallelly_1.35.0
[81] spatstat.random_3.1-5 beachmat_2.14.2
[83] scales_1.2.1 magrittr_2.0.3
[85] plyr_1.8.8 ica_1.0-3
[87] zlibbioc_1.44.0 compiler_4.2.2
[89] dqrng_0.3.0 clue_0.3-64
[91] fitdistrplus_1.1-11 snakecase_0.11.0
[93] cli_3.6.1 XVector_0.38.0
[95] listenv_0.9.0 pbapply_1.7-0
[97] ps_1.7.5 MASS_7.3-60
[99] tidyselect_1.2.0 stringi_1.7.12
[101] highr_0.10 yaml_2.3.7
[103] BiocSingular_1.14.0 locfit_1.5-9.7
[105] ggrepel_0.9.3 sass_0.4.6
[107] tools_4.2.2 timechange_0.2.0
[109] future.apply_1.10.0 parallel_4.2.2
[111] circlize_0.4.15 rstudioapi_0.14
[113] foreach_1.5.2 git2r_0.32.0
[115] gridExtra_2.3 rmdformats_1.0.4
[117] farver_2.1.1 Rtsne_0.16
[119] digest_0.6.31 BiocManager_1.30.20
[121] shiny_1.7.4 Rcpp_1.0.10
[123] egg_0.4.5 later_1.3.1
[125] RcppAnnoy_0.0.20 httr_1.4.6
[127] naturalsort_0.1.3 colorspace_2.1-0
[129] fs_1.6.2 tensor_1.5
[131] reticulate_1.28 splines_4.2.2
[133] uwot_0.1.14 spatstat.utils_3.0-3
[135] sp_1.6-0 plotly_4.10.1
[137] xtable_1.8-4 jsonlite_1.8.4
[139] R6_2.5.1 pillar_1.9.0
[141] htmltools_0.5.5 mime_0.12
[143] glue_1.6.2 fastmap_1.1.1
[145] BiocParallel_1.32.6 BiocNeighbors_1.16.0
[147] codetools_0.2-19 utf8_1.2.3
[149] lattice_0.21-8 bslib_0.4.2
[151] spatstat.sparse_3.0-1 ggbeeswarm_0.7.2
[153] leiden_0.4.3 magick_2.7.4
[155] survival_3.5-5 limma_3.54.2
[157] rmarkdown_2.21 munsell_0.5.0
[159] GetoptLong_1.0.5 rhdf5_2.42.1
[161] GenomeInfoDbData_1.2.9 iterators_1.0.14
[163] HDF5Array_1.26.0 reshape2_1.4.4
[165] gtable_0.3.3