Preprocessing the C133_Neeland_batch0 data set
Jovana Maksimovic
2024-02-26
Last updated: 2024-02-26
Checks: 6 1
Knit directory: paed-inflammation-CITEseq/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of
the R Markdown file created these results, you’ll want to first commit
it to the Git repo. If you’re still working on the analysis, you can
ignore this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20240216) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 7701592. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Untracked files:
Untracked: .DS_Store
Untracked: analysis/02.0_quality_control.Rmd
Untracked: analysis/03.0_call_doublets.Rmd
Untracked: code/dropletutils.R
Untracked: code/utility.R
Untracked: data/.DS_Store
Untracked: data/C133_Neeland_batch0/
Untracked: data/C133_Neeland_batch1/
Untracked: data/C133_Neeland_batch2/
Untracked: data/C133_Neeland_batch3/
Untracked: data/C133_Neeland_batch4/
Untracked: data/C133_Neeland_batch5/
Untracked: data/C133_Neeland_batch6/
Untracked: renv.lock
Untracked: renv/
Unstaged changes:
Modified: .Rprofile
Modified: .gitignore
Modified: analysis/01.0_preprocess_batch0.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown
(analysis/01.0_preprocess_batch0.Rmd) and HTML
(docs/01.0_preprocess_batch0.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | ecfde3d | Jovana Maksimovic | 2024-02-26 | Build site. |
| Rmd | 025ceae | Jovana Maksimovic | 2024-02-26 | wflow_publish(c("analysis/index.Rmd", "analysis/01*")) |
| html | da8df16 | Jovana Maksimovic | 2024-02-26 | Build site. |
| Rmd | 00319de | Jovana Maksimovic | 2024-02-26 | wflow_publish("analysis/01.0_preprocess_batch0.Rmd") |
suppressPackageStartupMessages({
library(here)
library(BiocStyle)
library(ggplot2)
library(cowplot)
library(patchwork)
library(tidyverse)
library(SingleCellExperiment)
library(DropletUtils)
library(scater)
})Overview
Bronchoalveolar lavage (BAL) samples were collected from 4
individuals: 1 control sample and 3 cystic fibrosis (CF) samples. The
samples were run on teh 10X Chromium and sequenced at the Garvan-Weizmann
Centre for Cellular Genomics (GWCCG). The multiplexed samples were
sequenced on an Illumina NovaSeq 6000 (NovaSeq Control Software v1.3.1 /
Real Time Analysis v3.3.3) ) using a NovaSeq S1 200 cycle kit (Illumina,
20012864). The cellranger count pipeline (version 6.0.2)
was used for alignment, filtering, barcode counting, and UMI counting
from FASTQ files. The GRCh38 reference was used for the alignment. The
number of cells from the pipeline was forced to 10,000. View the
CellRanger capture-specific web summaries: A, B, C, D.
sample_metadata_df <- read_csv(
here("data/C133_Neeland_batch0/data/sample_sheets/Sample_information.csv"))
knitr::kable(sample_metadata_df)| Participant | Sample | Sex | Age | Disease |
|---|---|---|---|---|
| A1_Ctrl | C | M | 3.00 | Ctrl |
| B1_CF | A | M | 2.99 | CF |
| C1_CF | B | M | 2.99 | CF |
| D1_CF | D | M | 3.03 | CF |
Set up the data
sce <- readRDS(here("data", "C133_Neeland_batch0",
"data", "SCEs", "C133_Neeland_batch0.CellRanger.SCE.rds"))
sce$Capture <- factor(sce$Sample)
capture_names <- levels(sce$Capture)
capture_names <- setNames(capture_names, capture_names)
sce$Sample <- NULL
sceclass: SingleCellExperiment
dim: 33538 8853584
metadata(1): Samples
assays(1): counts
rownames(33538): ENSG00000243485 ENSG00000237613 ... ENSG00000277475
ENSG00000268674
rowData names(3): ID Symbol Type
colnames(8853584): 1_AAACCCAAGAAACACT-1 1_AAACCCAAGAAACCCG-1 ...
4_TTTGTTGTCTTTGGAG-1 4_TTTGTTGTCTTTGGCT-1
colData names(2): Barcode Capture
reducedDimNames(0):
mainExpName: Gene Expression
altExpNames(0):Call cells from empty droplets
par(mfrow = c(2, 2))
lapply(capture_names, function(cn) {
sce <- sce[, sce$Capture == cn]
bcrank <- barcodeRanks(counts(sce))
# Only showing unique points for plotting speed.
uniq <- !duplicated(bcrank$rank)
plot(
x = bcrank$rank[uniq],
y = bcrank$total[uniq],
log = "xy",
xlab = "Rank",
ylab = "Total UMI count",
main = cn,
cex.lab = 1.2,
xlim = c(1, 500000),
ylim = c(1, 200000))
abline(h = metadata(bcrank)$inflection, col = "darkgreen", lty = 2)
abline(h = metadata(bcrank)$knee, col = "dodgerblue", lty = 2)
})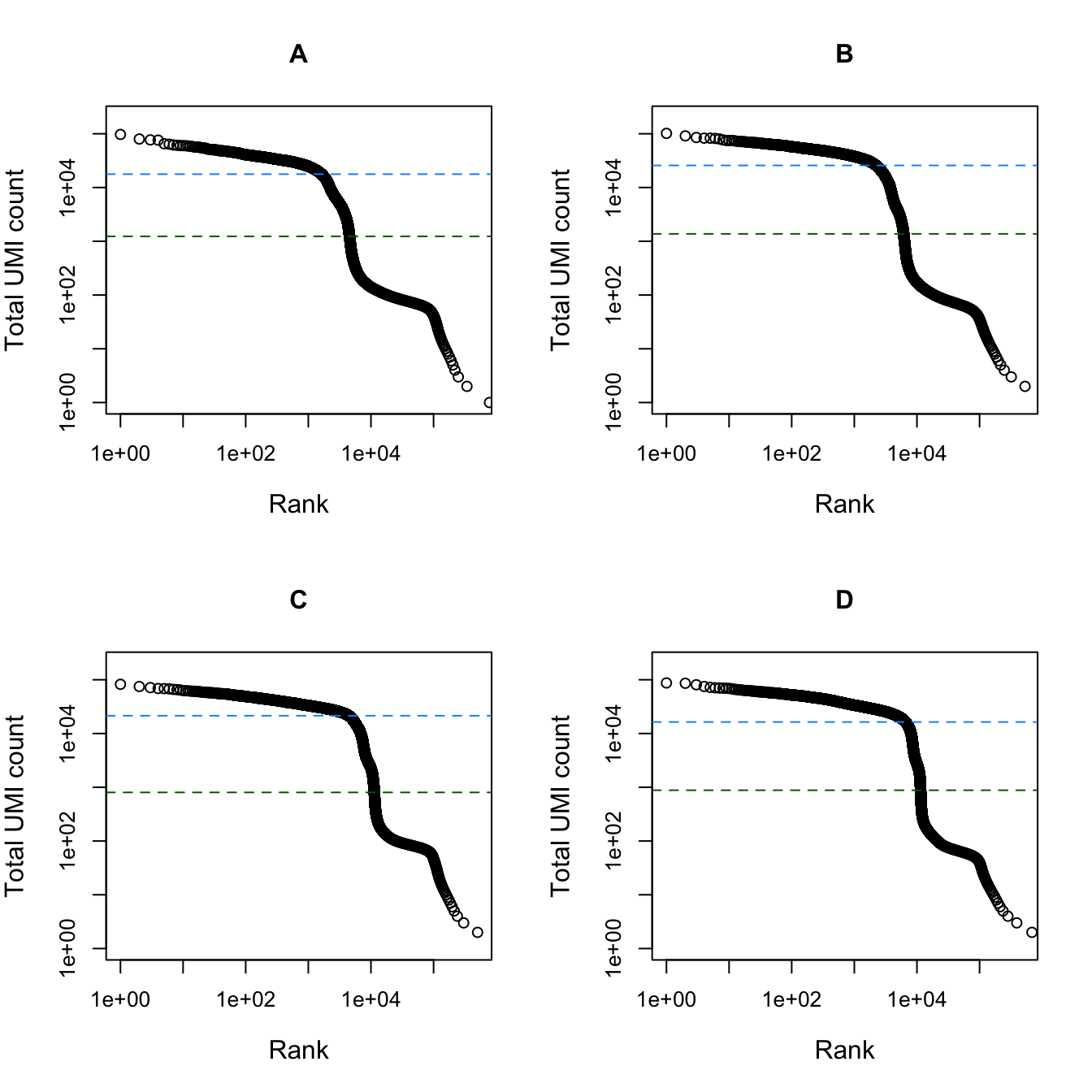
Total UMI count for each barcode in the dataset, plotted against its rank (in decreasing order of total counts). The inferred locations of the inflection (dark green dashed lines) and knee points (blue dashed lines) are also shown.
| Version | Author | Date |
|---|---|---|
| da8df16 | Jovana Maksimovic | 2024-02-26 |
Remove empty droplets.
empties <- do.call(rbind, lapply(capture_names, function(cn) {
message(cn)
empties <- readRDS(
here("data",
"C133_Neeland_batch0",
"data",
"emptyDrops", paste0(cn, ".emptyDrops.rds")))
empties$Capture <- cn
empties
}))
tapply(
empties$FDR,
empties$Capture,
function(x) sum(x <= 0.001, na.rm = TRUE)) |>
knitr::kable(
caption = "Number of non-empty droplets identified using `emptyDrops()` from **DropletUtils**.")| x | |
|---|---|
| A | 4980 |
| B | 6093 |
| C | 11197 |
| D | 12313 |
sce <- sce[, which(empties$FDR <= 0.001)]
sceclass: SingleCellExperiment
dim: 33538 34583
metadata(1): Samples
assays(1): counts
rownames(33538): ENSG00000243485 ENSG00000237613 ... ENSG00000277475
ENSG00000268674
rowData names(3): ID Symbol Type
colnames(34583): 1_AAACCCAAGCTAGTTC-1 1_AAACCCACAAGATTGA-1 ...
4_TTTGTTGTCTAGTACG-1 4_TTTGTTGTCTCGAACA-1
colData names(2): Barcode Capture
reducedDimNames(0):
mainExpName: Gene Expression
altExpNames(0):Add per cell quality control information
sce <- scuttle::addPerCellQC(sce)
head(colData(sce)) %>%
data.frame %>%
knitr::kable()| Barcode | Capture | sum | detected | total | |
|---|---|---|---|---|---|
| 1_AAACCCAAGCTAGTTC-1 | AAACCCAAGCTAGTTC-1 | A | 8126 | 2657 | 8126 |
| 1_AAACCCACAAGATTGA-1 | AAACCCACAAGATTGA-1 | A | 193 | 157 | 193 |
| 1_AAACCCACAGTCGCTG-1 | AAACCCACAGTCGCTG-1 | A | 22141 | 4536 | 22141 |
| 1_AAACCCAGTACCTAAC-1 | AAACCCAGTACCTAAC-1 | A | 17230 | 4084 | 17230 |
| 1_AAACCCATCGATTGGT-1 | AAACCCATCGATTGGT-1 | A | 32312 | 5092 | 32312 |
| 1_AAACCCATCGTCTCAC-1 | AAACCCATCGTCTCAC-1 | A | 28301 | 5044 | 28301 |
Save data
saveRDS(
sce,
here("data",
"C133_Neeland_batch0",
"data",
"SCEs",
"C133_Neeland_batch0.preprocessed.SCE.rds"))Session info
sessionInfo()R version 4.3.2 (2023-10-31)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS Sonoma 14.3
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: Australia/Melbourne
tzcode source: internal
attached base packages:
[1] stats4 stats graphics grDevices datasets utils methods
[8] base
other attached packages:
[1] scater_1.30.1 scuttle_1.12.0
[3] DropletUtils_1.22.0 SingleCellExperiment_1.24.0
[5] SummarizedExperiment_1.32.0 Biobase_2.62.0
[7] GenomicRanges_1.54.1 GenomeInfoDb_1.38.6
[9] IRanges_2.36.0 S4Vectors_0.40.2
[11] BiocGenerics_0.48.1 MatrixGenerics_1.14.0
[13] matrixStats_1.2.0 lubridate_1.9.3
[15] forcats_1.0.0 stringr_1.5.1
[17] dplyr_1.1.4 purrr_1.0.2
[19] readr_2.1.5 tidyr_1.3.1
[21] tibble_3.2.1 tidyverse_2.0.0
[23] patchwork_1.2.0 cowplot_1.1.3
[25] ggplot2_3.4.4 BiocStyle_2.30.0
[27] here_1.0.1 workflowr_1.7.1
loaded via a namespace (and not attached):
[1] bitops_1.0-7 gridExtra_2.3
[3] rlang_1.1.3 magrittr_2.0.3
[5] git2r_0.33.0 compiler_4.3.2
[7] getPass_0.2-4 DelayedMatrixStats_1.24.0
[9] callr_3.7.3 vctrs_0.6.5
[11] pkgconfig_2.0.3 crayon_1.5.2
[13] fastmap_1.1.1 XVector_0.42.0
[15] utf8_1.2.4 promises_1.2.1
[17] rmarkdown_2.25 tzdb_0.4.0
[19] ggbeeswarm_0.7.2 ps_1.7.6
[21] bit_4.0.5 xfun_0.42
[23] zlibbioc_1.48.0 cachem_1.0.8
[25] beachmat_2.18.1 jsonlite_1.8.8
[27] highr_0.10 later_1.3.2
[29] rhdf5filters_1.14.1 DelayedArray_0.28.0
[31] Rhdf5lib_1.24.2 BiocParallel_1.36.0
[33] irlba_2.3.5.1 parallel_4.3.2
[35] R6_2.5.1 bslib_0.6.1
[37] stringi_1.8.3 limma_3.58.1
[39] jquerylib_0.1.4 Rcpp_1.0.12
[41] knitr_1.45 R.utils_2.12.3
[43] httpuv_1.6.14 Matrix_1.6-5
[45] timechange_0.3.0 tidyselect_1.2.0
[47] viridis_0.6.5 rstudioapi_0.15.0
[49] abind_1.4-5 yaml_2.3.8
[51] codetools_0.2-19 processx_3.8.3
[53] lattice_0.22-5 withr_3.0.0
[55] evaluate_0.23 pillar_1.9.0
[57] BiocManager_1.30.22 whisker_0.4.1
[59] renv_1.0.3 generics_0.1.3
[61] vroom_1.6.5 rprojroot_2.0.4
[63] RCurl_1.98-1.14 hms_1.1.3
[65] sparseMatrixStats_1.14.0 munsell_0.5.0
[67] scales_1.3.0 glue_1.7.0
[69] tools_4.3.2 BiocNeighbors_1.20.2
[71] ScaledMatrix_1.10.0 locfit_1.5-9.8
[73] fs_1.6.3 rhdf5_2.46.1
[75] grid_4.3.2 edgeR_4.0.15
[77] colorspace_2.1-0 GenomeInfoDbData_1.2.11
[79] beeswarm_0.4.0 BiocSingular_1.18.0
[81] HDF5Array_1.30.0 vipor_0.4.7
[83] rsvd_1.0.5 cli_3.6.2
[85] fansi_1.0.6 viridisLite_0.4.2
[87] S4Arrays_1.2.0 gtable_0.3.4
[89] R.methodsS3_1.8.2 sass_0.4.8
[91] digest_0.6.34 ggrepel_0.9.5
[93] SparseArray_1.2.4 dqrng_0.3.2
[95] htmltools_0.5.7 R.oo_1.26.0
[97] lifecycle_1.0.4 httr_1.4.7
[99] statmod_1.5.0 bit64_4.0.5